

Cardiac hypertrophy is not amplified by deletion of cGMP-dependent protein kinase I in cardiomyocytes
- PMID: 20212138
- PMCID: PMC2851748
- DOI: 10.1073/pnas.1001360107
Cardiac hypertrophy is not amplified by deletion of cGMP-dependent protein kinase I in cardiomyocytes
Abstract
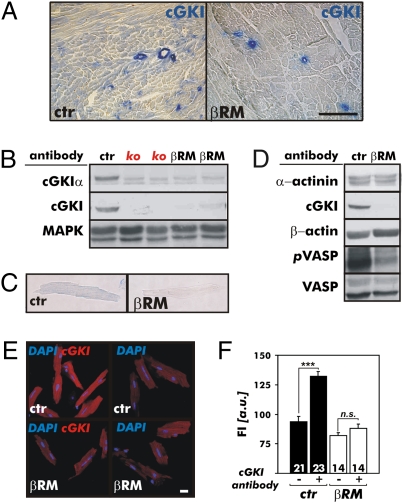
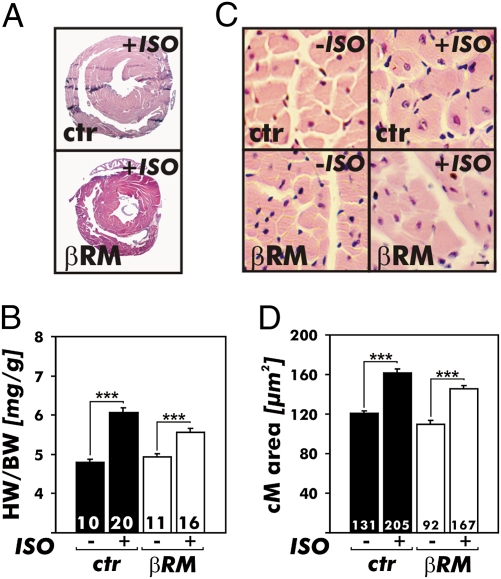
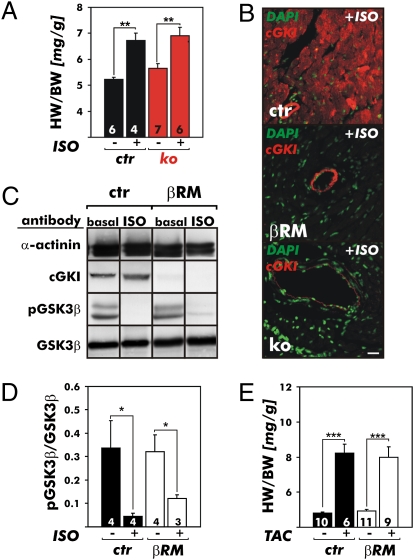
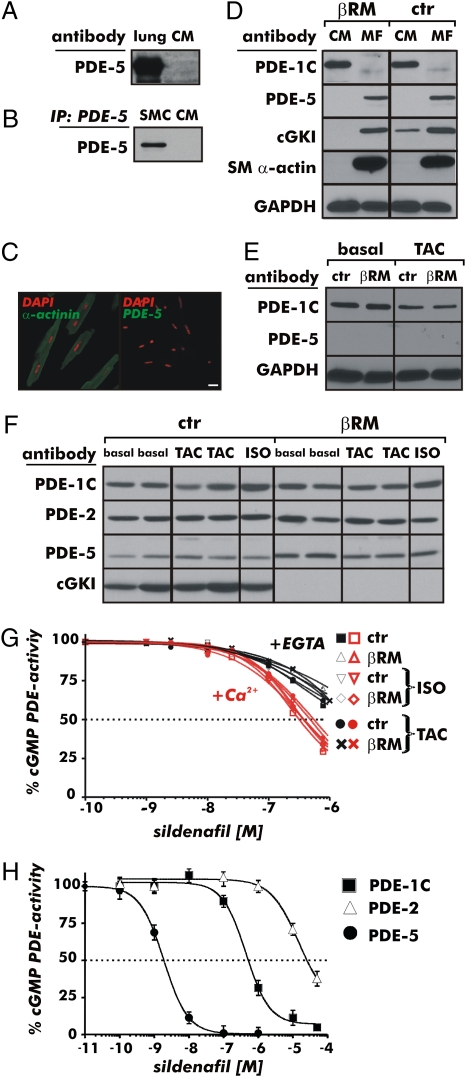
It has been suggested that cGMP kinase I (cGKI) dampens cardiac hypertrophy. We have compared the effect of isoproterenol (ISO) and transverse aortic constriction (TAC) on hypertrophy in WT [control (CTR)] mice, total cGKI-KO mice, and cGKIbeta rescue mice (betaRM) lacking cGKI specifically in cardiomyocytes (CMs). Infusion of ISO did not change the expression of cGKI in the hearts of CTR mice or betaRM but raised the heart weight by approximately 20% in both. An identical hypertrophic growth response was measured in CMs from CTR mice and betaRM and in isolated adult CMs cultured with or without 1 muM ISO. In both genotypes, ISO infusion induced similar changes in the expression of hypertrophy-associated cardiac genes and significant elevation of serum atrial natriuretic peptide and total cardiac cGMP. No differences in cardiac hypertrophy were obtained by 7-day ISO infusion in 4- to 6-week-old conventional cGKI-KO and CTR mice. Furthermore, TAC-induced hypertrophy of CTR mice and betaRM was not different and did not result in changes of the cGMP-hydrolyzing phosphodiesterase activities in hypertropic hearts or CMs. These results strongly suggest that cardiac myocyte cGKI does not affect the development of heart hypertrophy induced by pressure overload or chronic ISO infusion.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Comment in
-
Regulation and role of myocyte cyclic GMP-dependent protein kinase-1.Proc Natl Acad Sci U S A. 2010 Jun 15;107(24):E98; author reply E99. doi: 10.1073/pnas.1003889107. Epub 2010 May 24. Proc Natl Acad Sci U S A. 2010. PMID: 20498039 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
