

Channelopathies in Cav1.1, Cav1.3, and Cav1.4 voltage-gated L-type Ca2+ channels
- PMID: 20213496
- PMCID: PMC2883925
- DOI: 10.1007/s00424-010-0800-x
Channelopathies in Cav1.1, Cav1.3, and Cav1.4 voltage-gated L-type Ca2+ channels
Abstract
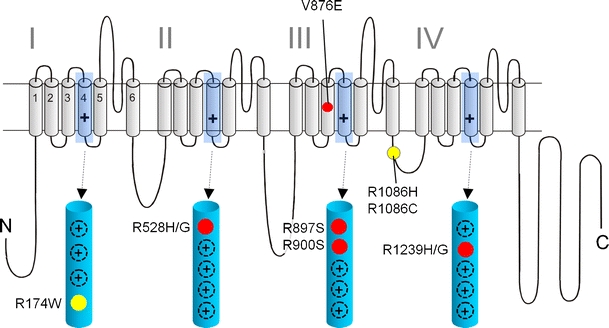
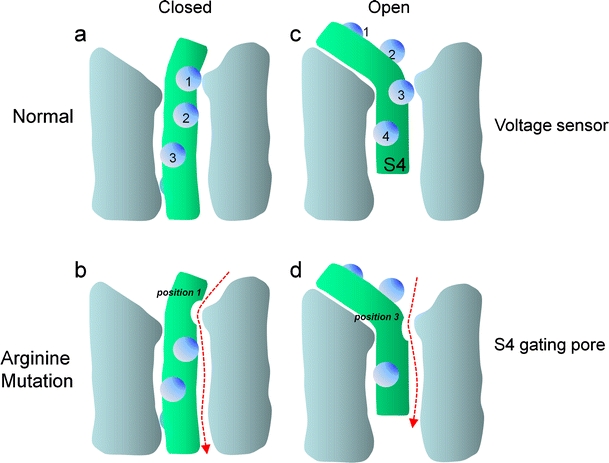
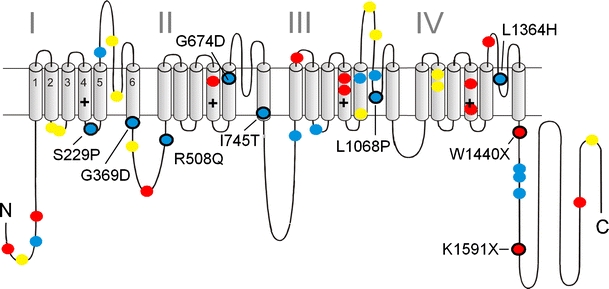
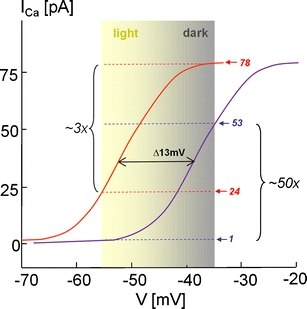
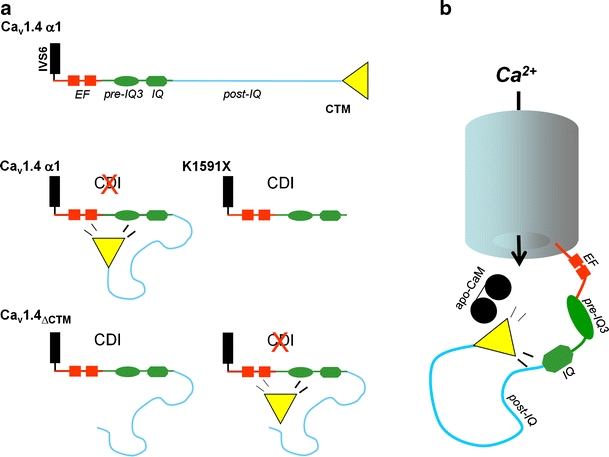
Voltage-gated Ca2+ channels couple membrane depolarization to Ca2+-dependent intracellular signaling events. This is achieved by mediating Ca2+ ion influx or by direct conformational coupling to intracellular Ca2+ release channels. The family of Cav1 channels, also termed L-type Ca2+ channels (LTCCs), is uniquely sensitive to organic Ca2+ channel blockers and expressed in many electrically excitable tissues. In this review, we summarize the role of LTCCs for human diseases caused by genetic Ca2+ channel defects (channelopathies). LTCC dysfunction can result from structural aberrations within their pore-forming alpha1 subunits causing hypokalemic periodic paralysis and malignant hyperthermia sensitivity (Cav1.1 alpha1), incomplete congenital stationary night blindness (CSNB2; Cav1.4 alpha1), and Timothy syndrome (Cav1.2 alpha1; reviewed separately in this issue). Cav1.3 alpha1 mutations have not been reported yet in humans, but channel loss of function would likely affect sinoatrial node function and hearing. Studies in mice revealed that LTCCs indirectly also contribute to neurological symptoms in Ca2+ channelopathies affecting non-LTCCs, such as Cav2.1 alpha1 in tottering mice. Ca2+ channelopathies provide exciting disease-related molecular detail that led to important novel insight not only into disease pathophysiology but also to mechanisms of channel function.
Figures

References
-
- Ball SL, Gregg RG. Using mutant mice to study the role of voltage-gated calcium channels in the retina. Adv Exp Med Biol. 2002;514:439–450. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
