

Metformin: a therapeutic opportunity in breast cancer
- PMID: 20215559
- PMCID: PMC2840206
- DOI: 10.1158/1078-0432.CCR-09-1805
Metformin: a therapeutic opportunity in breast cancer
Abstract
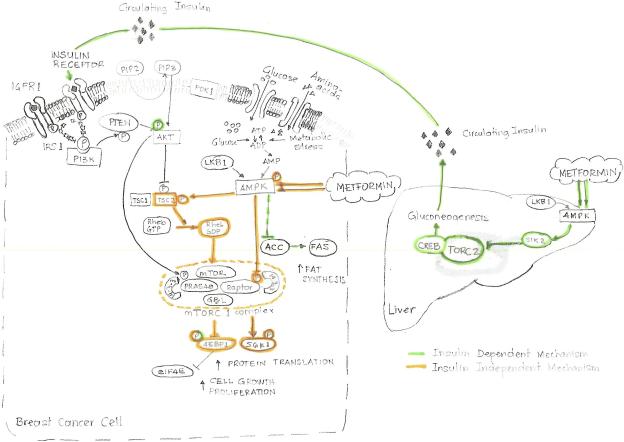
Two important, related pathways are involved in cancer growth: the insulin/insulin-like growth factor-1 (IGF1) signaling pathway, which is activated when nutrients are available, and the adenosine mono-phosphate-activated protein kinase (AMPK) pathway, activated when cells are starved for carbohydrates. Metformin inhibits transcription of key gluconeogenesis genes in the liver, increases glucose uptake in skeletal muscle, and decreases circulating insulin levels. Metformin reduces levels of circulating glucose, increases insulin sensitivity, and reduces insulin resistance-associated hyperinsulinemia. At the level of cell signaling, metformin activates AMPK. There are extensive preclinical data showing the anticancer effects of metformin in all breast cancer subtypes as well as in cytotoxic therapy-resistant models. These data, and the epidemiological and retrospective data supporting the antineoplastic effects of metformin, provide the rationale to study the role of metformin for breast cancer therapy in a variety of clinical settings.
Figures
Similar articles
-
Changes in insulin receptor signaling underlie neoadjuvant metformin administration in breast cancer: a prospective window of opportunity neoadjuvant study.Breast Cancer Res. 2015 Mar 3;17(1):32. doi: 10.1186/s13058-015-0540-0. Breast Cancer Res. 2015. PMID: 25849721 Free PMC article.
-
Metformin induces unique biological and molecular responses in triple negative breast cancer cells.Cell Cycle. 2009 Jul 1;8(13):2031-40. doi: 10.4161/cc.8.13.8814. Epub 2009 Jul 21. Cell Cycle. 2009. PMID: 19440038
-
Metformin and the mTOR inhibitor everolimus (RAD001) sensitize breast cancer cells to the cytotoxic effect of chemotherapeutic drugs in vitro.Anticancer Res. 2012 May;32(5):1627-37. Anticancer Res. 2012. PMID: 22593441
-
Anticancer effects of metformin and its potential use as a therapeutic agent for breast cancer.Future Oncol. 2011 Jun;7(6):727-36. doi: 10.2217/fon.11.49. Future Oncol. 2011. PMID: 21675836 Review.
-
Metformin: Insights into its anticancer potential with special reference to AMPK dependent and independent pathways.Life Sci. 2017 Sep 15;185:53-62. doi: 10.1016/j.lfs.2017.07.029. Epub 2017 Jul 26. Life Sci. 2017. PMID: 28755883 Review.
Cited by
-
Metformin represses self-renewal of the human breast carcinoma stem cells via inhibition of estrogen receptor-mediated OCT4 expression.PLoS One. 2011;6(11):e28068. doi: 10.1371/journal.pone.0028068. Epub 2011 Nov 23. PLoS One. 2011. PMID: 22132214 Free PMC article.
-
Efficacy of Metformin as Adjuvant Therapy in Metastatic Breast Cancer Treatment.J Clin Med. 2022 Sep 20;11(19):5505. doi: 10.3390/jcm11195505. J Clin Med. 2022. PMID: 36233373 Free PMC article.
-
Metformin: multi-faceted protection against cancer.Oncotarget. 2011 Dec;2(12):896-917. doi: 10.18632/oncotarget.387. Oncotarget. 2011. PMID: 22203527 Free PMC article. Review.
-
The C Allele of ATM rs11212617 Associates With Higher Pathological Complete Remission Rate in Breast Cancer Patients Treated With Neoadjuvant Metformin.Front Oncol. 2019 Mar 28;9:193. doi: 10.3389/fonc.2019.00193. eCollection 2019. Front Oncol. 2019. PMID: 30984619 Free PMC article.
-
Design and development of a peptide-based adiponectin receptor agonist for cancer treatment.BMC Biotechnol. 2011 Oct 5;11:90. doi: 10.1186/1472-6750-11-90. BMC Biotechnol. 2011. PMID: 21974986 Free PMC article.
References
-
- Towel MC, Hardie DG. AMP-Activated Protein Kinase in Metabolic Control and Insulin Signaling. Circulation Res. 2007;100:328–41. - PubMed
-
- Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–90. - PubMed
-
- Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–57. - PubMed
-
- Kovacic S, Soltys CL, Barr AJ, Shiojima I, Walsh K, Dyck JR. Akt activity negatively regulates phosphorylation of AMP-activated protein kinase in the heart. J Biol Chem. 2003;278:39422–27. - PubMed
-
- Horman S, Vertommen D, Heath R, Neumann D, Mouton V, Woods A, Schlattner U, Wallimann T, Carling D, Hue L, Rider MH. Insulin antagonizes ischemia-induced Thr172 phosphorylation of AMP-activated protein kinase alpha-subunits in heart via hierarchical phosphorylation of Ser485/491. J Biol Chem. 2006;281:5335–40. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
