

Treatment strategies and clinical trial design in ADPKD
- PMID: 20219622
- PMCID: PMC4127876
- DOI: 10.1053/j.ackd.2010.01.006
Treatment strategies and clinical trial design in ADPKD
Abstract
More frequent utilization and continuous improvement of imaging techniques has enhanced appreciation of the high phenotypic variability of autosomal dominant polycystic kidney disease, improved understanding of its natural history, and facilitated the observation of its structural progression. At the same time, identification of the PKD1 and PKD2 genes has provided clues to how the disease develops when they (genetic mechanisms) and their encoded proteins (molecular mechanisms) are disrupted. Interventions designed to rectify downstream effects of these disruptions have been examined in animal models, and some are currently tested in clinical trials. Efforts are underway to determine whether interventions capable to slow down, stop, or reverse structural progression of the disease will also prevent decline of renal function and improve clinically significant outcomes.
2010 National Kidney Foundation, Inc. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of Interest Statement: Dr. Torres is an investigator in clinical trials supported by Otsuka, Novartis, and Wyeth Pharmaceuticals.
Figures
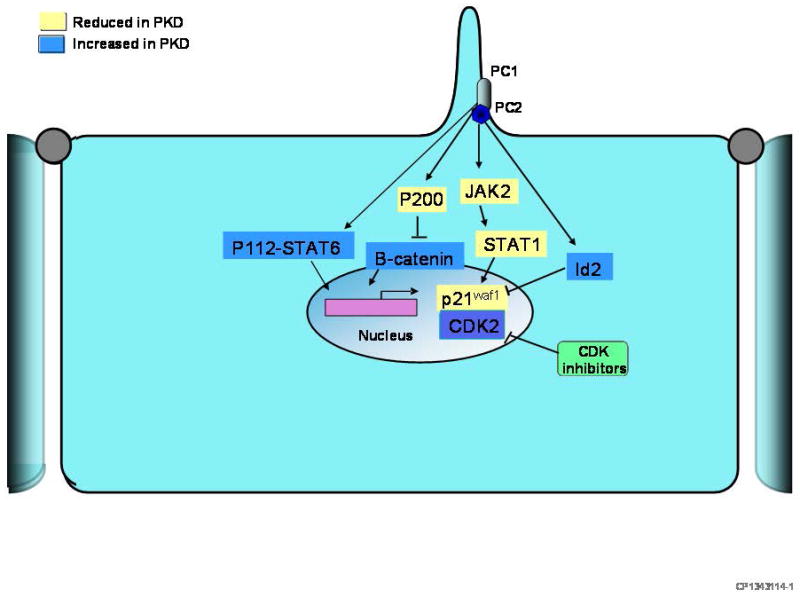
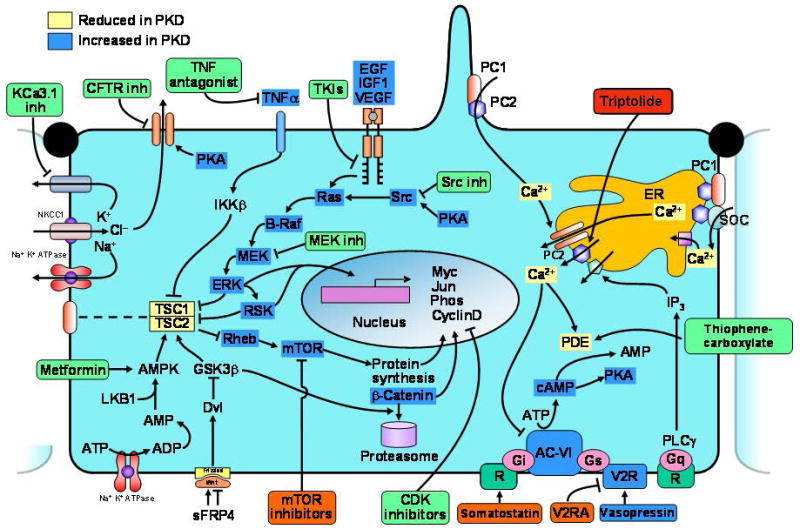
References
-
- Grantham JJ, Torres VE, Chapman AB, et al. Volume progression in polycystic kidney disease. N Engl J Med. 2006;354:2122–2130. - PubMed
-
- Kistler AD, Poster D, Krauer F, et al. Increases in kidney volume in autosomal dominant polycystic kidney disease can be detected within 6 months. Kidney Int. 2009;75(2):235–241. - PubMed
-
- Torres VE, King BF, Chapman AB, et al. Magnetic resonance measurements of renal blood flow and disease progression in autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2007;2(1):112–120. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
