

Sequence variability in clinical and laboratory isolates of herpes simplex virus 1 reveals new mutations
- PMID: 20219902
- PMCID: PMC2863834
- DOI: 10.1128/JVI.00312-10
Sequence variability in clinical and laboratory isolates of herpes simplex virus 1 reveals new mutations
Abstract
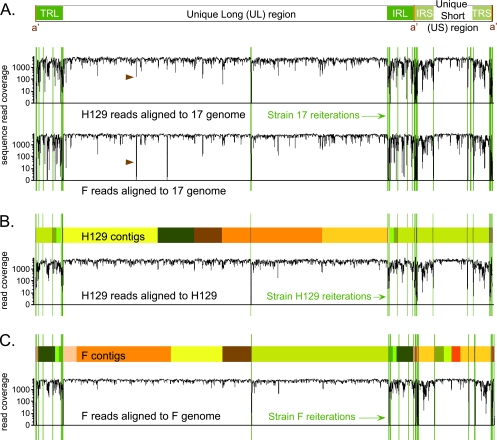
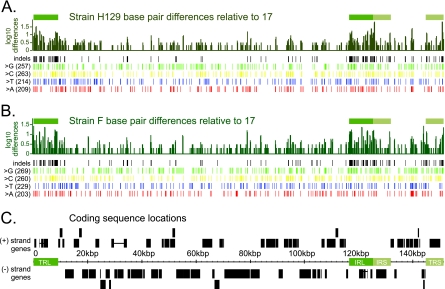
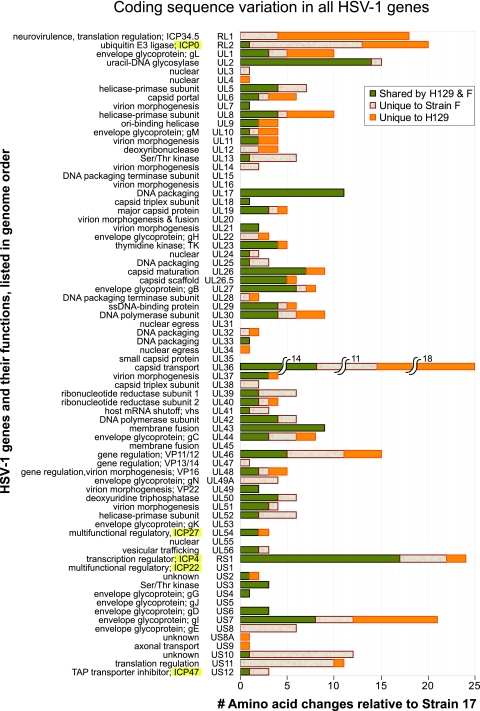
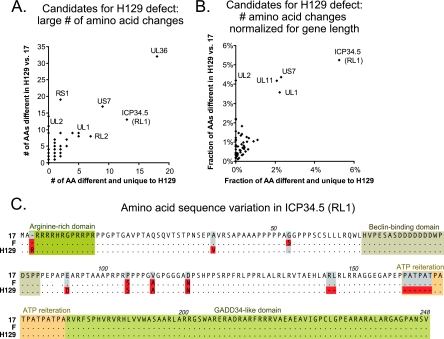
Herpes simplex virus 1 (HSV-1) is a well-adapted human pathogen that can invade the peripheral nervous system and persist there as a lifelong latent infection. Despite their ubiquity, only one natural isolate of HSV-1 (strain 17) has been sequenced. Using Illumina high-throughput sequencing of viral DNA, we obtained the genome sequences of both a laboratory strain (F) and a low-passage clinical isolate (H129). These data demonstrated the extent of interstrain variation across the entire genome of HSV-1 in both coding and noncoding regions. We found many amino acid differences distributed across the proteome of the new strain F sequence and the previously known strain 17, demonstrating the spectrum of variability among wild-type HSV-1 proteins. The clinical isolate, strain H129, displays a unique anterograde spread phenotype for which the causal mutations were completely unknown. We have defined the sequence differences in H129 and propose a number of potentially causal genes, including the neurovirulence protein ICP34.5 (RL1). Further studies will be required to demonstrate which change(s) is sufficient to recapitulate the spread defect of strain H129. Unexpectedly, these data also revealed a frameshift mutation in the UL13 kinase in our strain F isolate, demonstrating how deep genome sequencing can reveal the full complement of background mutations in any given strain, particularly those passaged or plaque purified in a laboratory setting. These data increase our knowledge of sequence variation in large DNA viruses and demonstrate the potential of deep sequencing to yield insight into DNA genome evolution and the variation among different pathogen isolates.
Figures
References
-
- Abaitua, F., R. N. Souto, H. Browne, T. Daikoku, and P. O'Hare. 2009. Characterization of the herpes simplex virus (HSV)-1 tegument protein VP1-2 during infection with the HSV temperature-sensitive mutant tsB7. J. Gen. Virol. 90:2353-2363. - PubMed
-
- Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403-410. - PubMed
-
- Archin, N. M., and S. S. Atherton. 2002. Rapid spread of a neurovirulent strain of HSV-1 through the CNS of BALB/c mice following anterior chamber inoculation. J. Neurovirol. 8:122-135. - PubMed
-
- Blondeau, C., N. Chbab, C. Beaumont, K. Courvoisier, N. Osterrieder, J. F. Vautherot, and C. Denesvre. 2007. A full UL13 open reading frame in Marek's disease virus (MDV) is dispensable for tumor formation and feather follicle tropism and cannot restore horizontal virus transmission of rRB-1B in vivo. Vet. Res. 38:419-433. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
