

Integrating multiple evidence sources to predict transcription factor binding in the human genome
- PMID: 20219943
- PMCID: PMC2847756
- DOI: 10.1101/gr.096305.109
Integrating multiple evidence sources to predict transcription factor binding in the human genome
Abstract
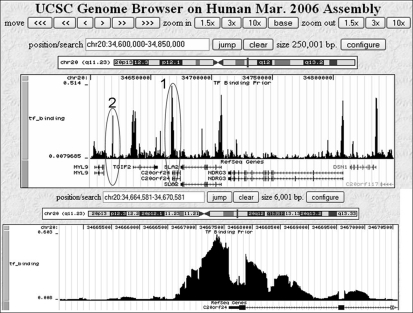
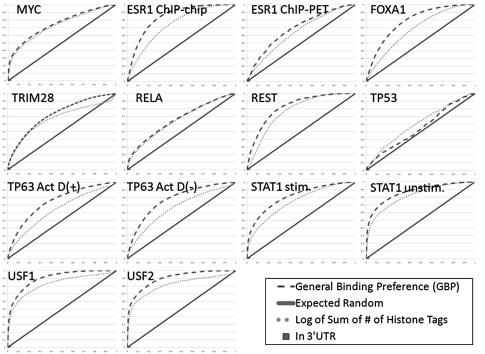
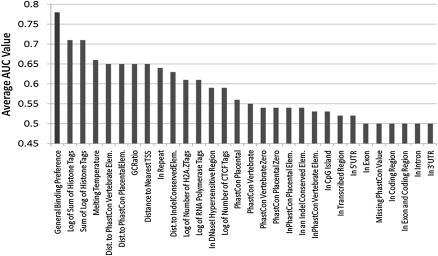
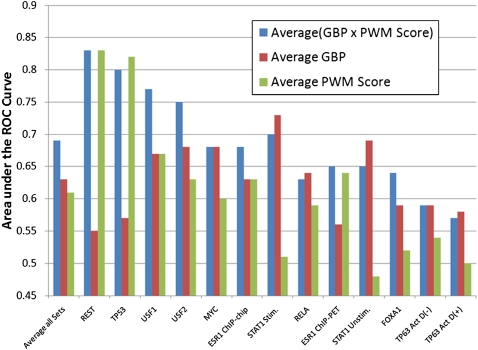
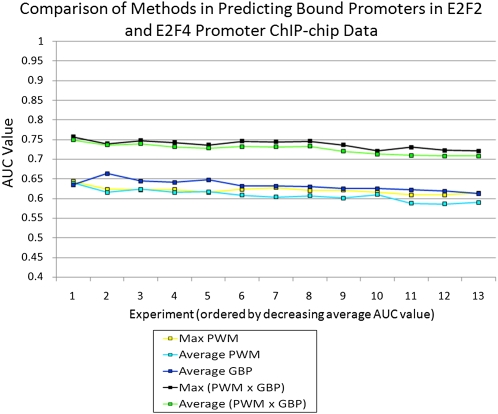
Information about the binding preferences of many transcription factors is known and characterized by a sequence binding motif. However, determining regions of the genome in which a transcription factor binds based on its motif is a challenging problem, particularly in species with large genomes, since there are often many sequences containing matches to the motif but are not bound. Several rules based on sequence conservation or location, relative to a transcription start site, have been proposed to help differentiate true binding sites from random ones. Other evidence sources may also be informative for this task. We developed a method for integrating multiple evidence sources using logistic regression classifiers. Our method works in two steps. First, we infer a score quantifying the general binding preferences of transcription factor binding at all locations based on a large set of evidence features, without using any motif specific information. Then, we combined this general binding preference score with motif information for specific transcription factors to improve prediction of regions bound by the factor. Using cross-validation and new experimental data we show that, surprisingly, the general binding preference can be highly predictive of true locations of transcription factor binding even when no binding motif is used. When combined with motif information our method outperforms previous methods for predicting locations of true binding.
Figures
References
-
- Bar-Joseph Z, Gerber G, Lee T, Rinaldi N, Yoo J, Robert F, Gordon B, Fraenkel E, Jaakkola T, Young R, et al. Computational discovery of gene modules and regulatory networks. Nat Biotechnol. 2003;21:1337–1342. - PubMed
-
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources