

Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer
- PMID: 20219944
- PMCID: PMC2847747
- DOI: 10.1101/gr.103606.109
Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer
Abstract
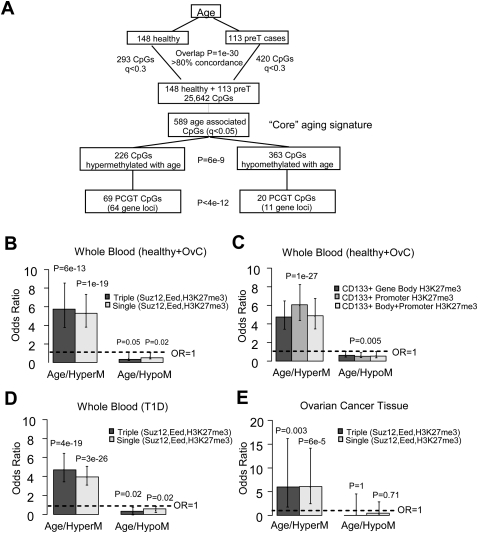
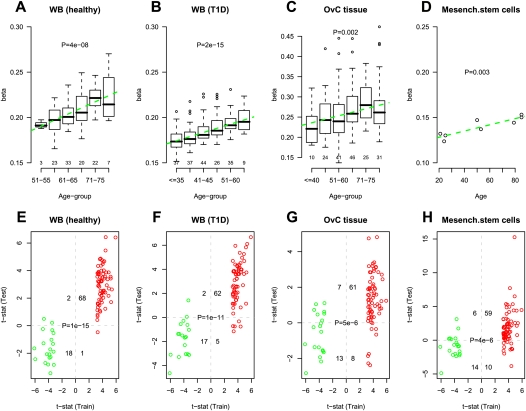
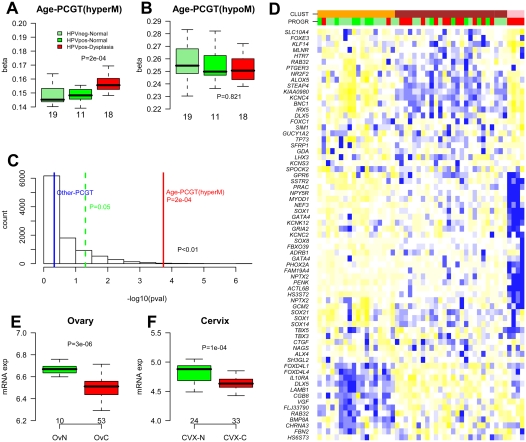
Polycomb group proteins (PCGs) are involved in repression of genes that are required for stem cell differentiation. Recently, it was shown that promoters of PCG target genes (PCGTs) are 12-fold more likely to be methylated in cancer than non-PCGTs. Age is the most important demographic risk factor for cancer, and we hypothesized that its carcinogenic potential may be referred by irreversibly stabilizing stem cell features. To test this, we analyzed the methylation status of over 27,000 CpGs mapping to promoters of approximately 14,000 genes in whole blood samples from 261 postmenopausal women. We demonstrate that stem cell PCGTs are far more likely to become methylated with age than non-targets (odds ratio = 5.3 [3.8-7.4], P < 10(-10)), independently of sex, tissue type, disease state, and methylation platform. We identified a specific subset of 69 PCGT CpGs that undergo hypermethylation with age and validated this methylation signature in seven independent data sets encompassing over 900 samples, including normal and cancer solid tissues and a population of bone marrow mesenchymal stem/stromal cells (P < 10(-5)). We find that the age-PCGT methylation signature is present in preneoplastic conditions and may drive gene expression changes associated with carcinogenesis. These findings shed substantial novel insights into the epigenetic effects of aging and support the view that age may predispose to malignant transformation by irreversibly stabilizing stem cell features.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases