

Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains
- PMID: 20219945
- PMCID: PMC2847746
- DOI: 10.1101/gr.103101.109
Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains
Abstract
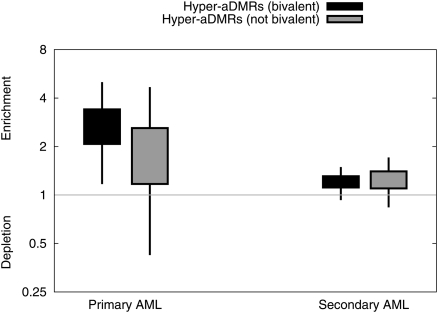
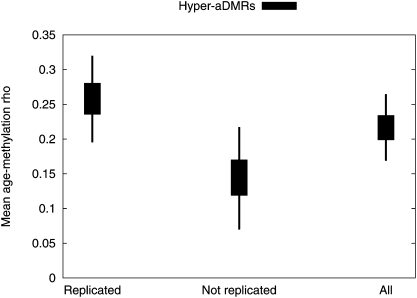
There is a growing realization that some aging-associated phenotypes/diseases have an epigenetic basis. Here, we report the first genome-scale study of epigenomic dynamics during normal human aging. We identify aging-associated differentially methylated regions (aDMRs) in whole blood in a discovery cohort, and then replicate these aDMRs in sorted CD4(+) T-cells and CD14(+) monocytes in an independent cohort, suggesting that aDMRs occur in precursor haematopoietic cells. Further replication of the aDMRs in buccal cells, representing a tissue that originates from a different germ layer compared with blood, demonstrates that the aDMR signature is a multitissue phenomenon. Moreover, we demonstrate that aging-associated DNA hypermethylation occurs predominantly at bivalent chromatin domain promoters. This same category of promoters, associated with key developmental genes, is frequently hypermethylated in cancers and in vitro cell culture, pointing to a novel mechanistic link between aberrant hypermethylation in cancer, aging, and cell culture.
Figures
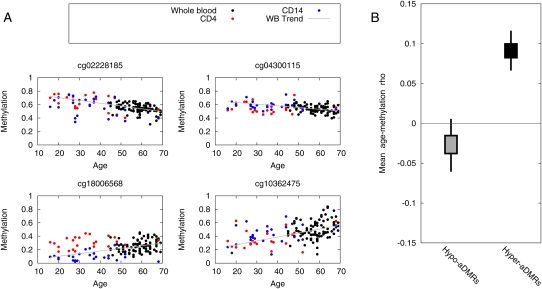
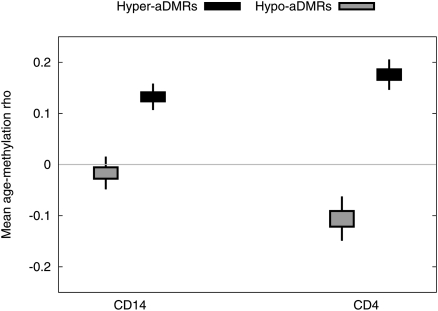
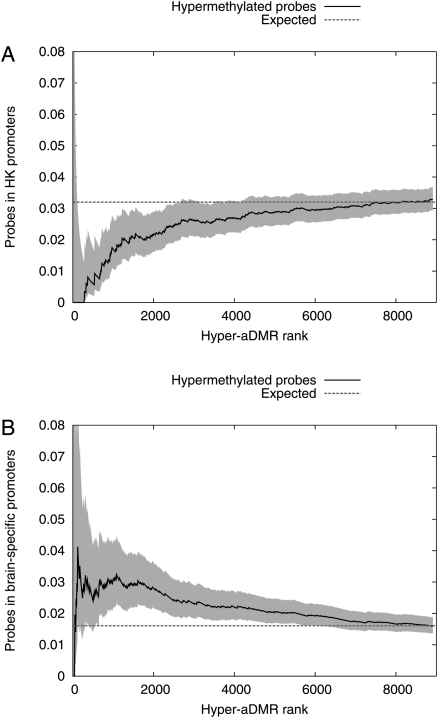
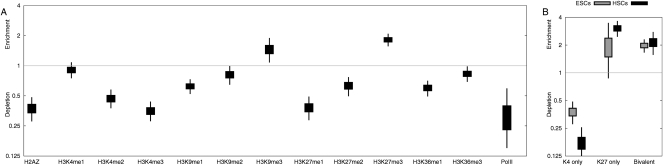
References
-
- Azuara V, Perry P, Sauer S, Spivakov M, Jørgensen HF, John RM, Gouti M, Casanova M, Warnes G, Merkenschlager M, et al. Chromatin signatures of pluripotent cell lines. Nat Cell Biol. 2006;8:532–538. - PubMed
-
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. - PubMed
-
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. - PubMed
-
- Bibikova M, Le J, Barnes R, Saedinia-Melnyk S, Zhou L, Shen R, Gunderson KL. Genome-wide DNA methylation profiling using Infinium® assay. Epigenomics. 2009;1:177–200. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials