

Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy
- PMID: 20220177
- PMCID: PMC4036802
- DOI: 10.1056/NEJMoa0908094
Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy
Abstract
Background: Whole-genome sequencing may revolutionize medical diagnostics through rapid identification of alleles that cause disease. However, even in cases with simple patterns of inheritance and unambiguous diagnoses, the relationship between disease phenotypes and their corresponding genetic changes can be complicated. Comprehensive diagnostic assays must therefore identify all possible DNA changes in each haplotype and determine which are responsible for the underlying disorder. The high number of rare, heterogeneous mutations present in all humans and the paucity of known functional variants in more than 90% of annotated genes make this challenge particularly difficult. Thus, the identification of the molecular basis of a genetic disease by means of whole-genome sequencing has remained elusive. We therefore aimed to assess the usefulness of human whole-genome sequencing for genetic diagnosis in a patient with Charcot-Marie-Tooth disease.
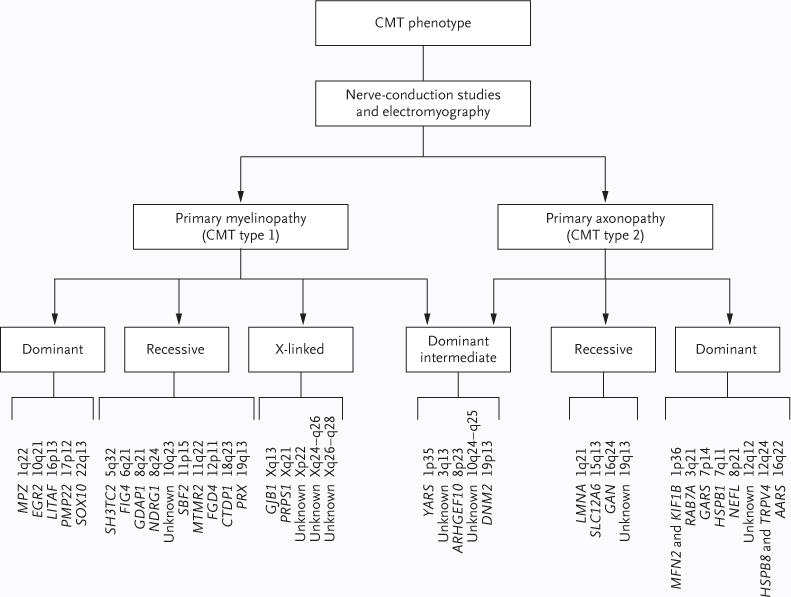
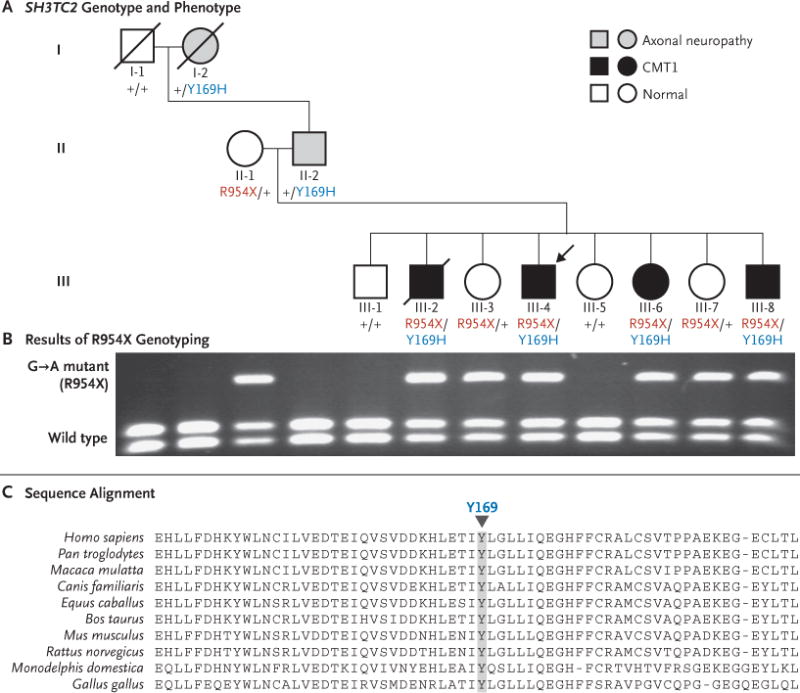
Methods: We identified a family with a recessive form of Charcot-Marie-Tooth disease for which the genetic basis had not been identified. We sequenced the whole genome of the proband, identified all potential functional variants in genes likely to be related to the disease, and genotyped these variants in the affected family members.
Results: We identified and validated compound, heterozygous, causative alleles in SH3TC2 (the SH3 domain and tetratricopeptide repeats 2 gene), involving two mutations, in the proband and in family members affected by Charcot-Marie-Tooth disease. Separate subclinical phenotypes segregated independently with each of the two mutations; heterozygous mutations confer susceptibility to neuropathy, including the carpal tunnel syndrome.
Conclusions: As shown in this study of a family with Charcot-Marie-Tooth disease, whole-genome sequencing can identify clinically relevant variants and provide diagnostic information to inform the care of patients.
2010 Massachusetts Medical Society
Conflict of interest statement
Disclosure forms provided by the authors are available with the full text of this article at
Figures
Comment in
-
Individual genomes on the horizon.N Engl J Med. 2010 Apr 1;362(13):1235-6. doi: 10.1056/NEJMe1001090. Epub 2010 Mar 10. N Engl J Med. 2010. PMID: 20220178 No abstract available.
References
-
- Rommens JM, Iannuzzi MC, Kerem B, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245:1059–65. - PubMed
-
- Wallace MR, Marchuk DA, Andersen LB, et al. Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients. Science. 1990;249:181–6. - PubMed
-
- Kajiwara K, Berson EL, Dryja TP. Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science. 1994;264:1604–8. - PubMed
-
- Katsanis N, Ansley SJ, Badano JL, et al. Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder. Science. 2001;293:2256–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases