

Mislocalization of XPF-ERCC1 nuclease contributes to reduced DNA repair in XP-F patients
- PMID: 20221251
- PMCID: PMC2832669
- DOI: 10.1371/journal.pgen.1000871
Mislocalization of XPF-ERCC1 nuclease contributes to reduced DNA repair in XP-F patients
Abstract
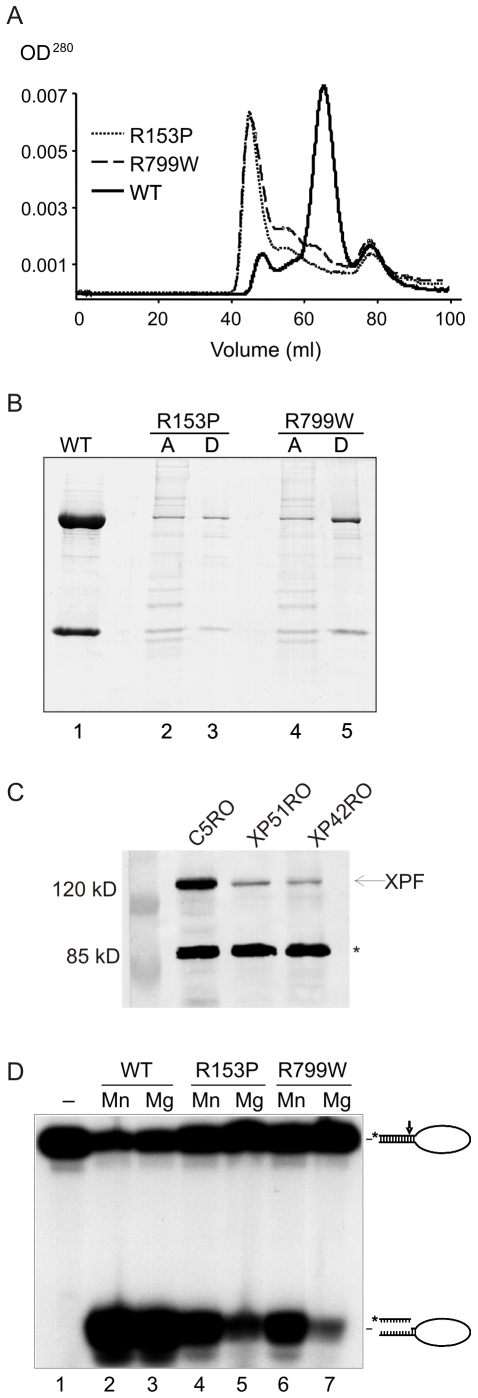
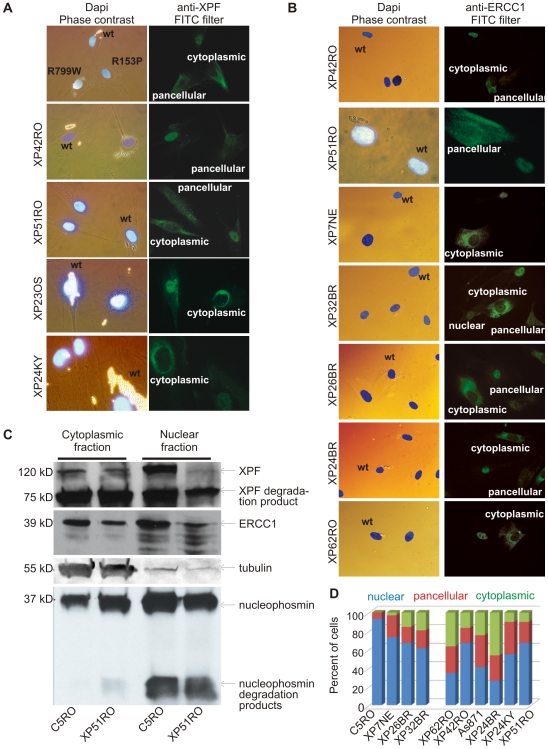
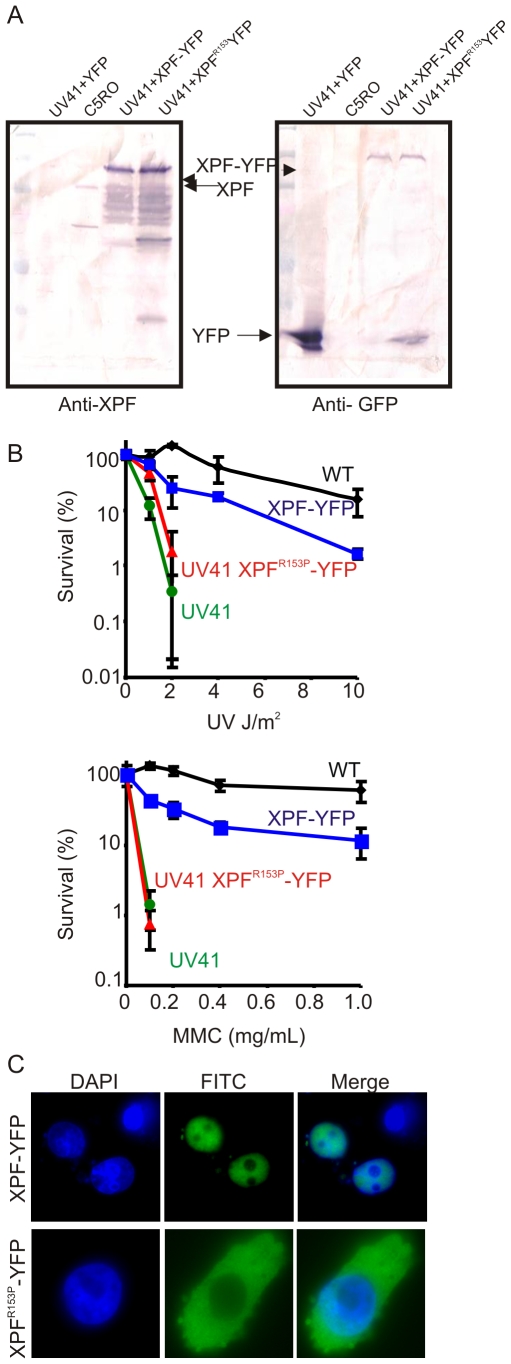
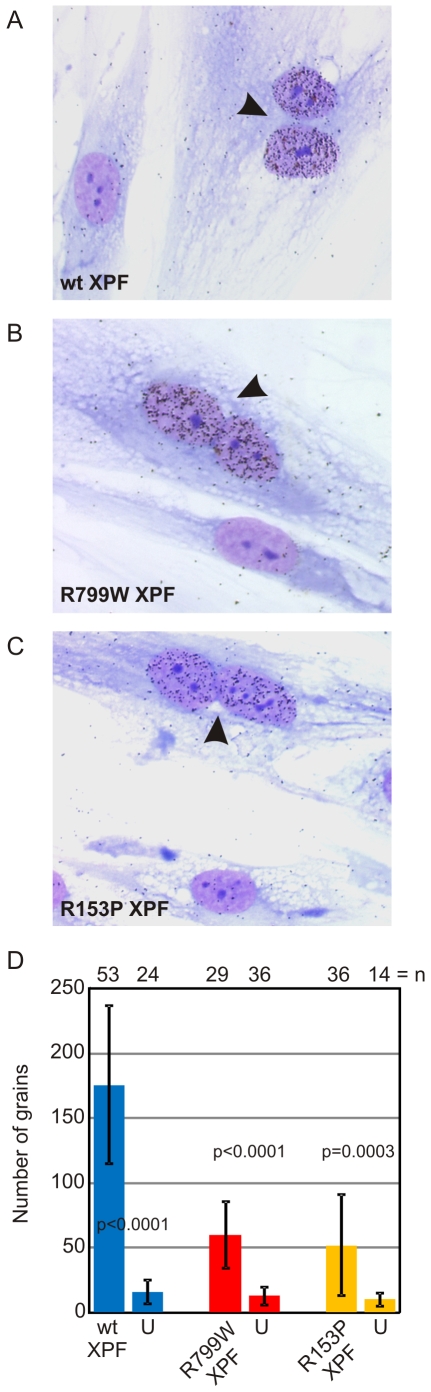
Xeroderma pigmentosum (XP) is caused by defects in the nucleotide excision repair (NER) pathway. NER removes helix-distorting DNA lesions, such as UV-induced photodimers, from the genome. Patients suffering from XP exhibit exquisite sun sensitivity, high incidence of skin cancer, and in some cases neurodegeneration. The severity of XP varies tremendously depending upon which NER gene is mutated and how severely the mutation affects DNA repair capacity. XPF-ERCC1 is a structure-specific endonuclease essential for incising the damaged strand of DNA in NER. Missense mutations in XPF can result not only in XP, but also XPF-ERCC1 (XFE) progeroid syndrome, a disease of accelerated aging. In an attempt to determine how mutations in XPF can lead to such diverse symptoms, the effects of a progeria-causing mutation (XPF(R153P)) were compared to an XP-causing mutation (XPF(R799W)) in vitro and in vivo. Recombinant XPF harboring either mutation was purified in a complex with ERCC1 and tested for its ability to incise a stem-loop structure in vitro. Both mutant complexes nicked the substrate indicating that neither mutation obviates catalytic activity of the nuclease. Surprisingly, differential immunostaining and fractionation of cells from an XFE progeroid patient revealed that XPF-ERCC1 is abundant in the cytoplasm. This was confirmed by fluorescent detection of XPF(R153P)-YFP expressed in Xpf mutant cells. In addition, microinjection of XPF(R153P)-ERCC1 into the nucleus of XPF-deficient human cells restored nucleotide excision repair of UV-induced DNA damage. Intriguingly, in all XPF mutant cell lines examined, XPF-ERCC1 was detected in the cytoplasm of a fraction of cells. This demonstrates that at least part of the DNA repair defect and symptoms associated with mutations in XPF are due to mislocalization of XPF-ERCC1 into the cytoplasm of cells, likely due to protein misfolding. Analysis of these patient cells therefore reveals a novel mechanism to potentially regulate a cell's capacity for DNA repair: by manipulating nuclear localization of XPF-ERCC1.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Xeroderma pigmentosum group F protein binds to Eg5 and is required for proper mitosis: implications for XP-F and XFE.Genes Cells. 2012 Mar;17(3):173-85. doi: 10.1111/j.1365-2443.2012.01582.x. Genes Cells. 2012. PMID: 22353549
-
Removal of reactive oxygen species-induced 3'-blocked ends by XPF-ERCC1.Chem Res Toxicol. 2011 Nov 21;24(11):1876-81. doi: 10.1021/tx200221j. Epub 2011 Oct 18. Chem Res Toxicol. 2011. PMID: 22007867 Free PMC article.
-
The XPA-binding domain of ERCC1 is required for nucleotide excision repair but not other DNA repair pathways.J Biol Chem. 2010 Feb 5;285(6):3705-3712. doi: 10.1074/jbc.M109.067538. Epub 2009 Nov 23. J Biol Chem. 2010. PMID: 19940136 Free PMC article.
-
Multiple roles of the ERCC1-XPF endonuclease in DNA repair and resistance to anticancer drugs.Anticancer Res. 2010 Sep;30(9):3223-32. Anticancer Res. 2010. PMID: 20944091 Review.
-
Xeroderma pigmentosum and molecular cloning of DNA repair genes.Anticancer Res. 1996 Mar-Apr;16(2):693-708. Anticancer Res. 1996. PMID: 8687116 Review.
Cited by
-
Age-related motor neuron degeneration in DNA repair-deficient Ercc1 mice.Acta Neuropathol. 2010 Oct;120(4):461-75. doi: 10.1007/s00401-010-0715-9. Epub 2010 Jul 4. Acta Neuropathol. 2010. PMID: 20602234 Free PMC article.
-
Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia.Am J Hum Genet. 2013 May 2;92(5):800-6. doi: 10.1016/j.ajhg.2013.04.002. Epub 2013 Apr 25. Am J Hum Genet. 2013. PMID: 23623386 Free PMC article.
-
The clinical spectrum associated with ERCC5 mutations: Is there a relationship between phenotype and genotype?Pediatr Discov. 2024 Jul 1;2(4):e71. doi: 10.1002/pdi3.71. eCollection 2024 Dec. Pediatr Discov. 2024. PMID: 40626125 Free PMC article.
-
Deficient expression of DNA repair enzymes in early progression to sporadic colon cancer.Genome Integr. 2012 Apr 11;3(1):3. doi: 10.1186/2041-9414-3-3. Genome Integr. 2012. PMID: 22494821 Free PMC article.
-
DNA repair and genomic stability in lungs affected by acute injury.Biomed Pharmacother. 2019 Nov;119:109412. doi: 10.1016/j.biopha.2019.109412. Epub 2019 Sep 9. Biomed Pharmacother. 2019. PMID: 31514069 Free PMC article. Review.
References
-
- Kraemer KH, Lee MM, Scotto J. Xeroderma pigmentosum. Cutaneous, ocular, and neurologic abnormalities in 830 published cases. Arch Dermatol. 1987;123:241–250. - PubMed
-
- Nouspikel T. Nucleotide excision repair and neurological diseases. DNA Repair (Amst) 2008;7:1155–1167. - PubMed
-
- Robbins JH, Brumback RA, Mendiones M, Barrett SF, Carl JR, et al. Neurological disease in xeroderma pigmentosum. Documentation of a late onset type of the juvenile onset form. Brain. 1991;114 ( Pt 3):1335–1361. - PubMed
-
- Gillet LC, Scharer OD. Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem Rev. 2006;106:253–276. - PubMed
-
- Sugasawa K, Ng JM, Masutani C, Iwai S, van der Spek PJ, et al. Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol Cell. 1998;2:223–232. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
