

Diffusion, crowding & protein stability in a dynamic molecular model of the bacterial cytoplasm
- PMID: 20221255
- PMCID: PMC2832674
- DOI: 10.1371/journal.pcbi.1000694
Diffusion, crowding & protein stability in a dynamic molecular model of the bacterial cytoplasm
Abstract
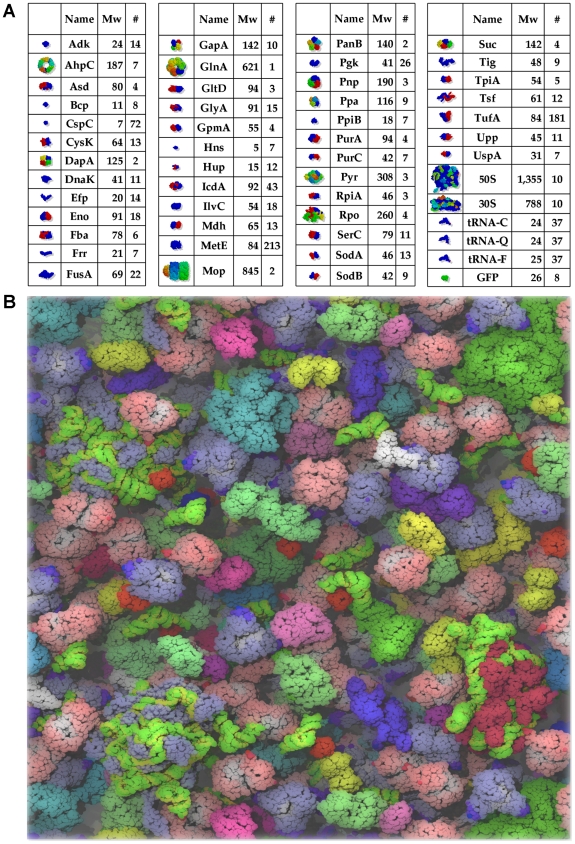
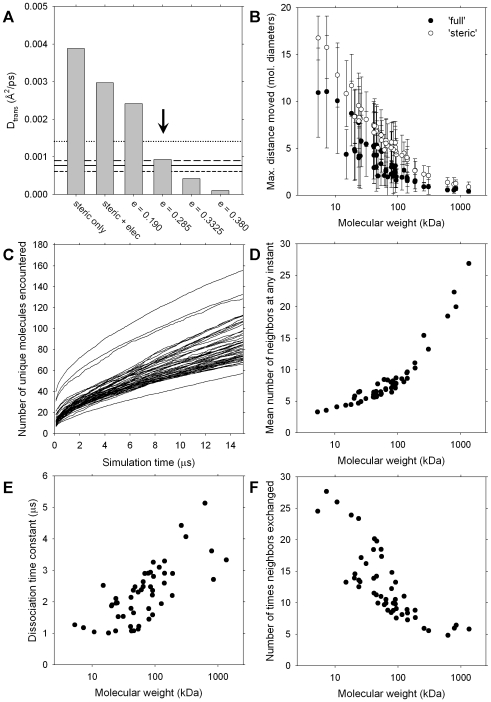
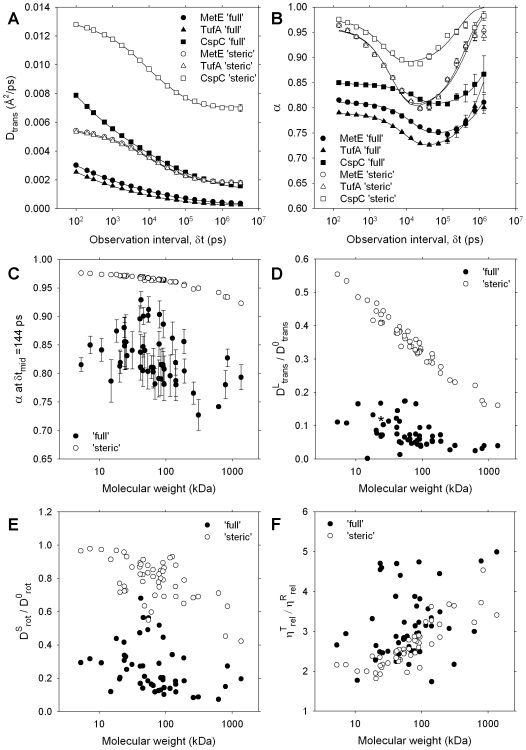
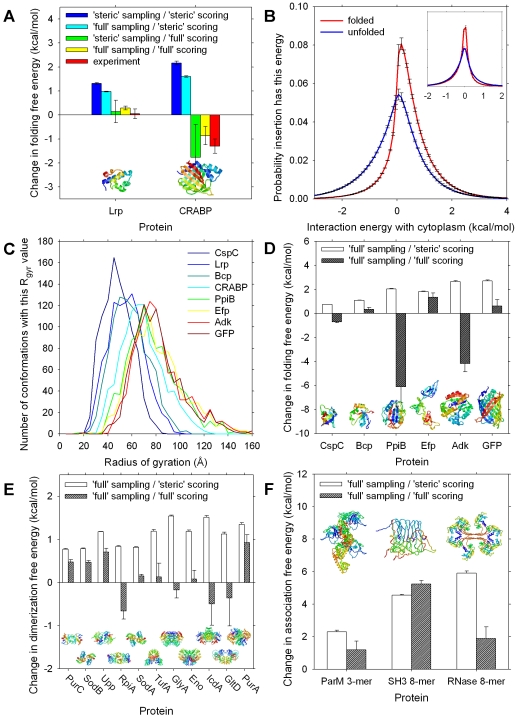
A longstanding question in molecular biology is the extent to which the behavior of macromolecules observed in vitro accurately reflects their behavior in vivo. A number of sophisticated experimental techniques now allow the behavior of individual types of macromolecule to be studied directly in vivo; none, however, allow a wide range of molecule types to be observed simultaneously. In order to tackle this issue we have adopted a computational perspective, and, having selected the model prokaryote Escherichia coli as a test system, have assembled an atomically detailed model of its cytoplasmic environment that includes 50 of the most abundant types of macromolecules at experimentally measured concentrations. Brownian dynamics (BD) simulations of the cytoplasm model have been calibrated to reproduce the translational diffusion coefficients of Green Fluorescent Protein (GFP) observed in vivo, and "snapshots" of the simulation trajectories have been used to compute the cytoplasm's effects on the thermodynamics of protein folding, association and aggregation events. The simulation model successfully describes the relative thermodynamic stabilities of proteins measured in E. coli, and shows that effects additional to the commonly cited "crowding" effect must be included in attempts to understand macromolecular behavior in vivo.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Dix JA, Verkman AS. Crowding effects on diffusion in solutions and cells. Annu Rev Biophys. 2008;37:247–263. - PubMed
-
- Zimmerman SB, Minton AP. Macromolecular crowding – biochemical, biophysical, and physiological consequences. Annu Rev Biophys Biomol Struct. 1993;22:27–65. - PubMed
-
- Ghaemmaghami S, Oas TG. Quantitative protein stability measurement in vivo. Nature Struct Biol. 2001;8:879–882. - PubMed
-
- Reckel S, Hänsel R, Löhr F, Dötsch V. In-cell NMR spectroscopy. Prog Nuc Mag Res Spec. 2007;51:91–101.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
