

Fatal transmissible amyloid encephalopathy: a new type of prion disease associated with lack of prion protein membrane anchoring
- PMID: 20221436
- PMCID: PMC2832701
- DOI: 10.1371/journal.ppat.1000800
Fatal transmissible amyloid encephalopathy: a new type of prion disease associated with lack of prion protein membrane anchoring
Abstract
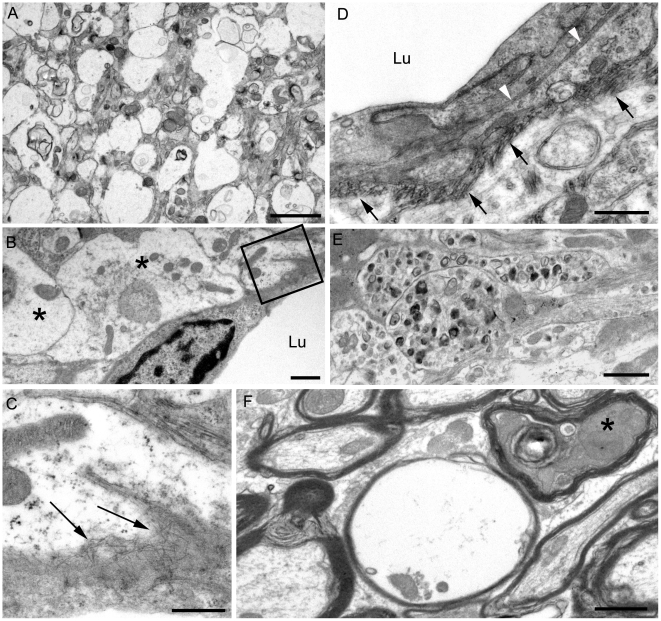
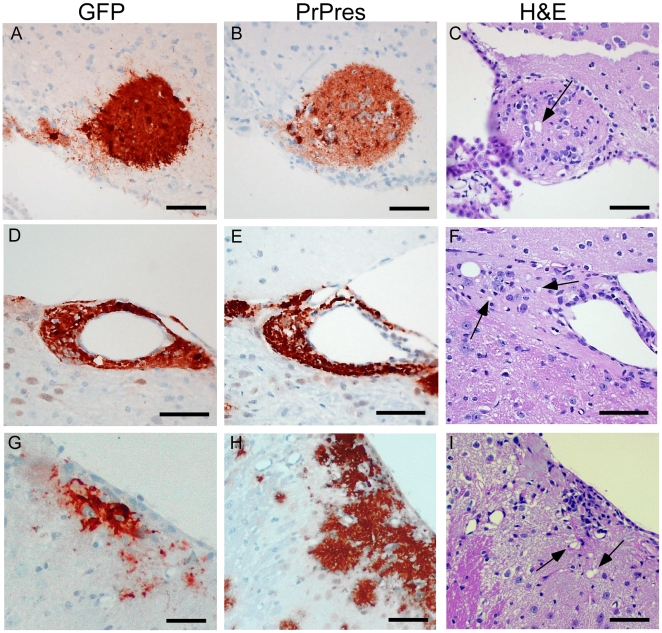
Prion diseases are fatal neurodegenerative diseases of humans and animals characterized by gray matter spongiosis and accumulation of aggregated, misfolded, protease-resistant prion protein (PrPres). PrPres can be deposited in brain in an amyloid-form and/or non-amyloid form, and is derived from host-encoded protease-sensitive PrP (PrPsen), a protein normally anchored to the plasma membrane by glycosylphosphatidylinositol (GPI). Previously, using heterozygous transgenic mice expressing only anchorless PrP, we found that PrP anchoring to the cell membrane was required for typical clinical scrapie. However, in the present experiments, using homozygous transgenic mice expressing two-fold more anchorless PrP, scrapie infection induced a new fatal disease with unique clinical signs and altered neuropathology, compared to non-transgenic mice expressing only anchored PrP. Brain tissue of transgenic mice had high amounts of infectivity, and histopathology showed dense amyloid PrPres plaque deposits without gray matter spongiosis. In contrast, infected non-transgenic mice had diffuse non-amyloid PrPres deposits with significant gray matter spongiosis. Brain graft studies suggested that anchored PrPsen expression was required for gray matter spongiosis during prion infection. Furthermore, electron and light microscopic studies in infected transgenic mice demonstrated several pathogenic processes not seen in typical prion disease, including cerebral amyloid angiopathy and ultrastructural alterations in perivascular neuropil. These findings were similar to certain human familial prion diseases as well as to non-prion human neurodegenerative diseases, such as Alzheimer's disease.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures





References
-
- Aguzzi A, Polymenidou M. Mammalian prion biology: one century of evolving concepts. Cell. 2004;116:313–327. - PubMed
-
- Di Bari MA, Chianini F, Vaccari G, Esposito E, Conte M, et al. The bank vole (Myodes glareolus) as a sensitive bioassay for sheep scrapie. J Gen Virol. 2008;89:2975–2985. - PubMed
-
- Nonno R, Di Bari MA, Cardone F, Vaccari G, Fazzi P, et al. Efficient transmission and characterization of Creutzfeldt-Jakob disease strains in bank voles. PLoS Pathog. 2006;2:e12. doi: 10.1371/journal.ppat.0020012. - DOI - PMC - PubMed
-
- Jeffrey M, Goodsir CM, Bruce ME, McBride PA, Fraser JR. In vivo toxicity of prion protein in murine scrapie: ultrastructural and immunogold studies. Neuropathol Appl Neurobiol. 1997;23:93–101. - PubMed
-
- Gonzalez L, Martin S, Begara-McGorum I, Hunter N, Houston F, et al. Effects of agent strain and host genotype on PrP accumulation in the brain of sheep naturally and experimentally affected with scrapie. J Comp Pathol. 2002;126:17–29. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
