

Decoding pathogenesis of slow-channel congenital myasthenic syndromes using recombinant expression and mice models
- PMID: 20222328
- PMCID: PMC2929179
Decoding pathogenesis of slow-channel congenital myasthenic syndromes using recombinant expression and mice models
Abstract
Despite the fact that they are orphan diseases, congenital myasthenic syndromes (CMS) challenge those who suffer from it by causing fatigable muscle weakness, in the most benign cases, to a progressive wasting of muscles that may sentence patients to a wheelchair or even death. Compared to other more common neurological diseases, CMS are rare. Nevertheless, extensive research in CMS is performed in laboratories such as ours. Among the diverse neuromuscular disorders of CMS, we are focusing in the slow-channel congenital myasthenic syndrome (SCS), which is caused by mutations in genes encoding acetylcholine receptor subunits. The study of SCS has evolved from clinical electrophysiological studies to in vitro expression systems and transgenic mice models. The present review evaluates the methodological approaches that are most commonly employed to assess synaptic impairment in SCS and also provides perspectives for new approaches. Electrophysiological methodologies typically employed by physicians to diagnose patients include electromyography, whereas patient muscle samples are used for intracellular recordings, single-channel recordings and toxin binding experiments. In vitro expression systems allow the study of a particular mutation without the need of patient intervention. Indeed, in vitro expression systems have usually been implicated in the development of therapeutic strategies such as quinidine- and fluoxetine-based treatments and, more recently, RNA interference. A breakthrough in the study of SCS has been the development of transgenic mice bearing the mutations that cause SCS. These transgenic mice models have actually been key in the elucidation of the pathogenesis of the SCS mutations by linking IP-3 receptors to calcium overloading, as well as caspases and calpains to the hallmark of SCS, namely endplate myopathy. Finally, we summarize our experiences with suspected SCS patients from a local perspective and comment on one aspect of the contribution of our group in the study of SCS.
Conflict of interest statement
The authors have no conflict of interest to disclose.
Figures
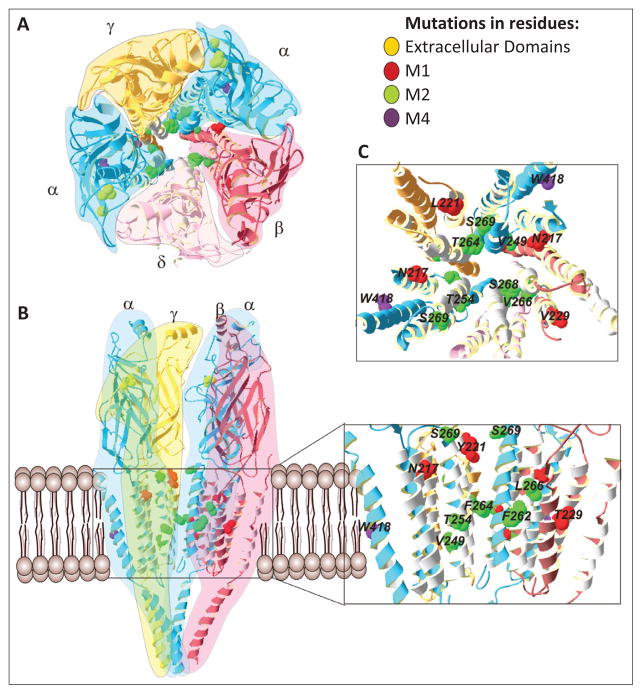
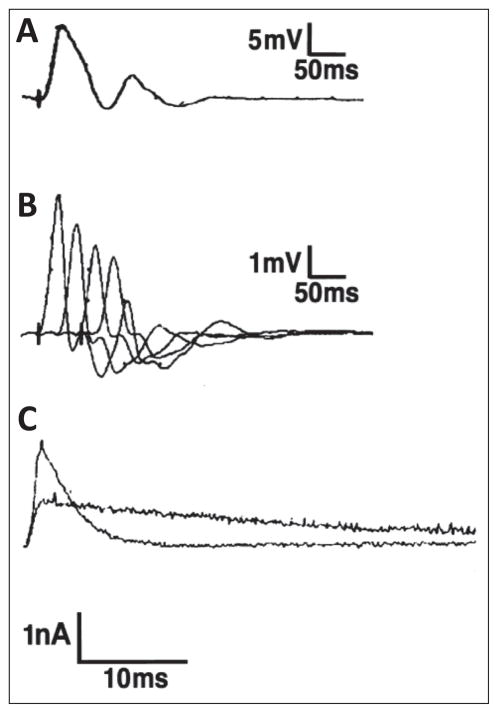
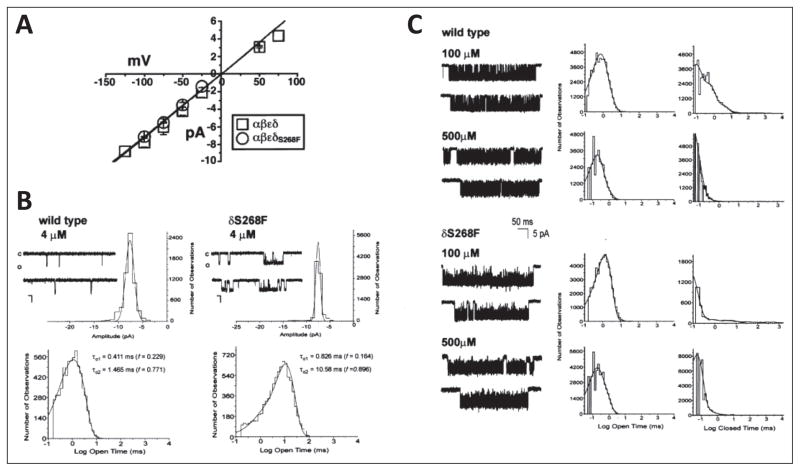
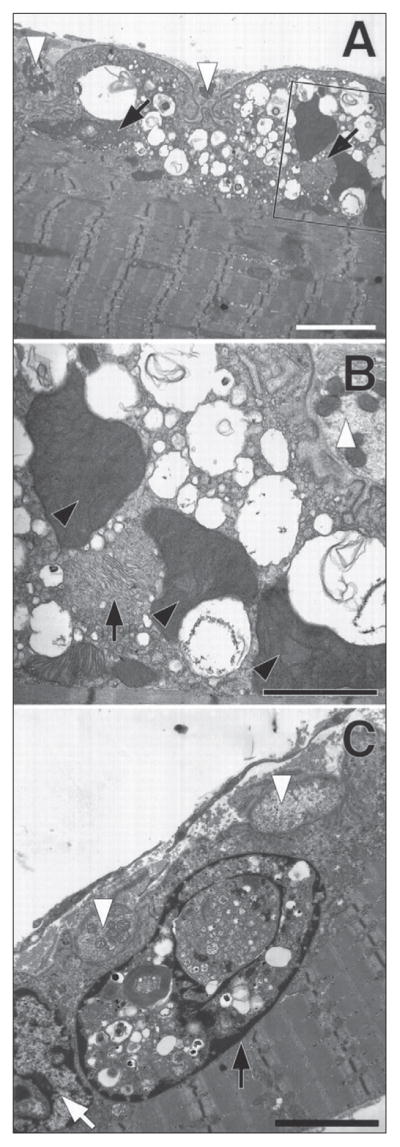
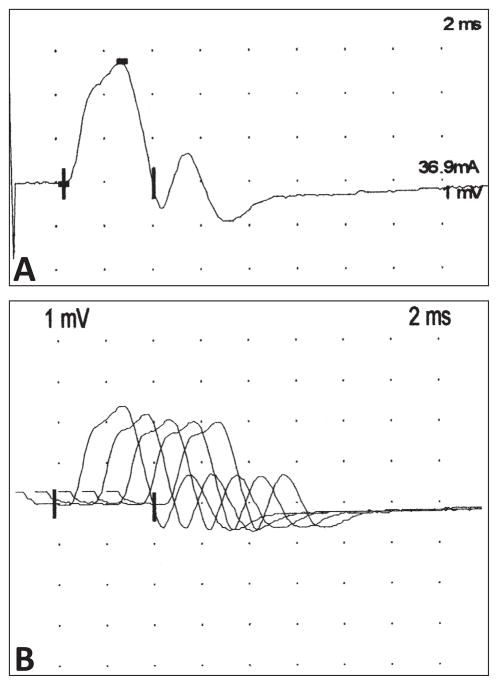
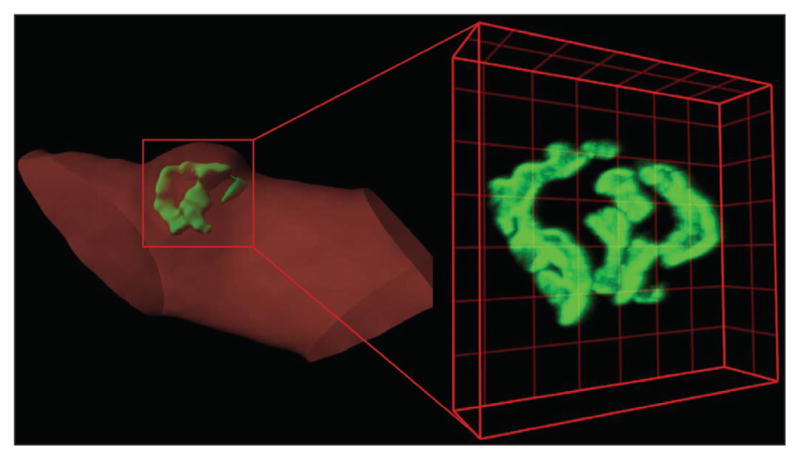
Similar articles
-
Active calcium accumulation underlies severe weakness in a panel of mice with slow-channel syndrome.J Neurosci. 2002 Aug 1;22(15):6447-57. doi: 10.1523/JNEUROSCI.22-15-06447.2002. J Neurosci. 2002. PMID: 12151524 Free PMC article.
-
A mouse model of the slow channel myasthenic syndrome: Neuromuscular physiology and effects of ephedrine treatment.Exp Neurol. 2013 Oct;248:286-98. doi: 10.1016/j.expneurol.2013.06.012. Epub 2013 Jun 21. Exp Neurol. 2013. PMID: 23797154
-
[Congenital myasthenic syndromes: phenotypic expression and pathophysiological characterisation].Rev Neurol (Paris). 2004 Feb;160(2):163-76. doi: 10.1016/s0035-3787(04)70887-5. Rev Neurol (Paris). 2004. PMID: 15034473 Review. French.
-
Novel delta subunit mutation in slow-channel syndrome causes severe weakness by novel mechanisms.Ann Neurol. 2002 Jan;51(1):102-12. doi: 10.1002/ana.10077. Ann Neurol. 2002. PMID: 11782989 Free PMC article.
-
Congenital Myasthenic Syndromes or Inherited Disorders of Neuromuscular Transmission: Recent Discoveries and Open Questions.J Neuromuscul Dis. 2017;4(4):269-284. doi: 10.3233/JND-170257. J Neuromuscul Dis. 2017. PMID: 29125502 Free PMC article. Review.
Cited by
-
Point Mutations of Nicotinic Receptor α1 Subunit Reveal New Molecular Features of G153S Slow-Channel Myasthenia.Molecules. 2021 Feb 26;26(5):1278. doi: 10.3390/molecules26051278. Molecules. 2021. PMID: 33652901 Free PMC article.
-
Makaluvamine G from the Marine Sponge Zyzzia fuliginosa Inhibits Muscle nAChR by Binding at the Orthosteric and Allosteric Sites.Mar Drugs. 2018 Mar 28;16(4):109. doi: 10.3390/md16040109. Mar Drugs. 2018. PMID: 29597332 Free PMC article.
-
Pharmacological Strategy for Congenital Myasthenic Syndrome with CHRNE Mutations: A Meta-Analysis of Case Reports.Curr Neuropharmacol. 2021;19(5):718-729. doi: 10.2174/1570159X18666200729092332. Curr Neuropharmacol. 2021. PMID: 32727330 Free PMC article.
-
Transgenic mouse model reveals an unsuspected role of the acetylcholine receptor in statin-induced neuromuscular adverse drug reactions.Pharmacogenomics J. 2013 Aug;13(4):362-8. doi: 10.1038/tpj.2012.21. Epub 2012 Jun 12. Pharmacogenomics J. 2013. PMID: 22688219 Free PMC article.
References
-
- Engel AG, Ohno K, Sine SM. Congenital myasthenic syndromes: recent advances. Arch Neurol. 1999;56:163–167. - PubMed
-
- Hantai D, Richard P, Koenig J, Eymard B. Congenital myasthenic syndromes. Curr Opin Neurol. 2004;17:539–551. - PubMed
-
- Harper CM. Congenital myasthenic syndromes. Semin Neurol. 2004;24:111–123. - PubMed
-
- Engel AG. Congenital myasthenic syndromes. Neurol Clin. 1994;12:401–437. - PubMed
-
- Engel AG, Lambert EH, Mulder DM, et al. A newly recognized congenital myasthenic syndrome attributed to a prolonged open time of the acetylcholine-induced ion channel. Ann Neurol. 1982;11:553–569. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources