

Review
doi: 10.1016/j.tem.2010.01.009.
Epub 2010 Mar 10.
Gluttony, sloth and the metabolic syndrome: a roadmap to lipotoxicity
Affiliations
- PMID: 20223680
- PMCID: PMC2880185
- DOI: 10.1016/j.tem.2010.01.009
Item in Clipboard
Review
Gluttony, sloth and the metabolic syndrome: a roadmap to lipotoxicity
Trends Endocrinol Metab.
2010 Jun.
Abstract
Once considered divine retribution for sins, comorbidities of obesity (metabolic syndrome) are today attributed to obesity-induced metabolic defects. Here, we propose that obesity and hyperleptinemia protect lipid-intolerant nonadipose organs against lipotoxic lipid spillover during sustained caloric surplus. Metabolic syndrome is ascribed to lipotoxicity caused by age-related resistance to antilipotoxic protection by leptin.
Published by Elsevier Ltd.
Figures
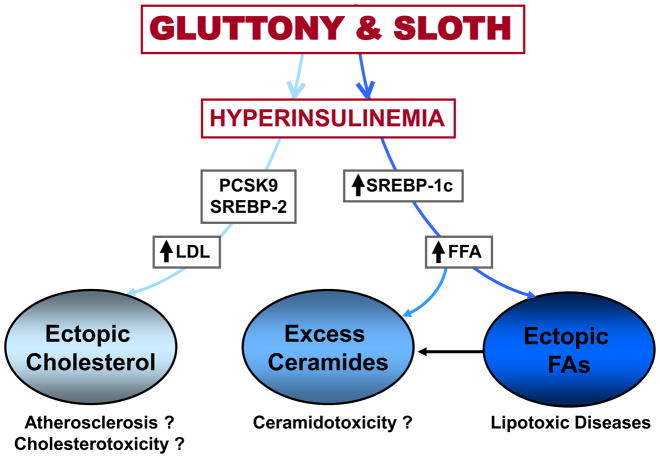
A biological explanation for the morbid and mortal consequences of a gluttonous and slothful lifestyle, implicating nutrient-induced upregulation of lipogenesis, cholesterologenesis and ceramidogenesis to the point of lipotoxicity. It should be pointed out that cholesterolotoxicity has not been demonstrated to occur spontaneously, but could exist (?) based on diabetes caused by transgenic overexpression of SREBP2 in β-cells [48]. Ceramidotoxicity, a potentially important metabolic aberration [49], is also not clearly identified in metabolic syndrome, but has been demonstrated to occur spontaneously in islets of ZDF rats with T2DM and metabolic syndrome [38].
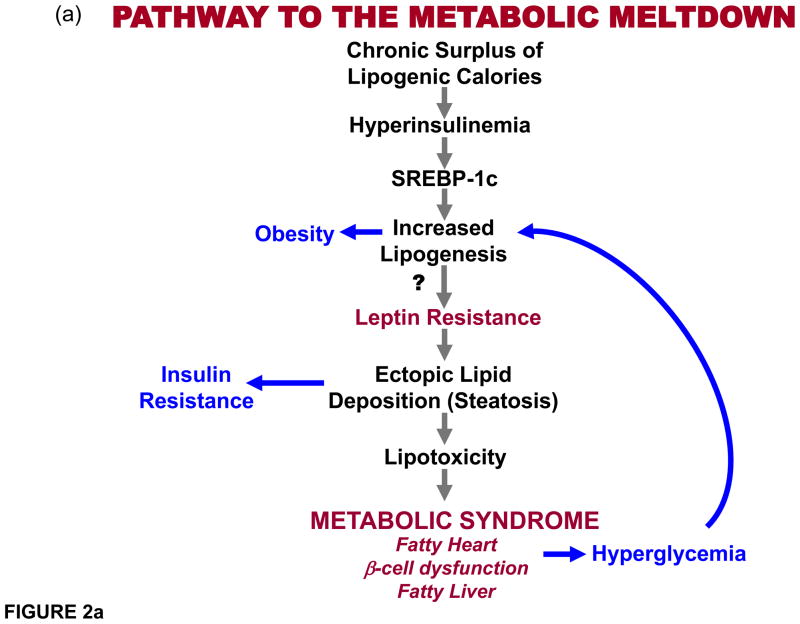
(a) Proposed pathway depicting the pathway to the metabolic syndrome. Chronic caloric surplus is here promoted to be the sine qua non for all subsequent events. It differs from more conventional views of the etiology of this disorder in three main respects: 1) Obesity, commonly considered a “disease” that causes metabolic syndrome, is depicted here as a normal physiologic response to caloric surplus that actually protects, at least temporarily, by sequestering toxic fatty acids in adipocytes that would otherwise damage organs. 2) Insulin resistance is also taken out of the etiologic mainstream to become a consequence, rather than cause, of the ectopic lipid deposition. 3) Leptin resistance is placed in a key causal role to explain why the hyperleptinemia of chronic overnutrition ultimately loses its ability to prevent ectopic lipid accumulation in target organs, at which point the metabolic syndrome is present. (b) Role of obesity-induced hyperleptinemia. Hyperleptinemia lowers fat content in peripheral organs. As adipocytes expand with triglycerides, leptin secretion increases proportionately. Since leptin stimulates fatty acid oxidation, adipocytes would be oxidizing, rather than storing, fat if the endogenous leptin they secrete were to act on them. Such an autocrine/paracrine relationship between the secretory product, leptin, and the secreting cells, is prevented by a progressive decline of leptin receptor expression. This physiologic leptin resistance is essential to permit accumulation of surplus calories into adipocytes. Meanwhile, the lipo-oxidative action of leptin is fully operative on peripheral organs, which minimizes ectopic lipid accumulation, at least temporarily. However, later in life peripheral organs also become leptin resistant. Leptin action on the hypothalamus limits the level of overnutrition without inhibiting it, while leptin’s lipooxidative action on the peripheral tissues keeps them free of ectopic lipid accumulation resulting from adipocyte spillover. If the disappearance of the leptin receptor during overnutrition is prevented by transgenic overexpression, obesity is prevented.
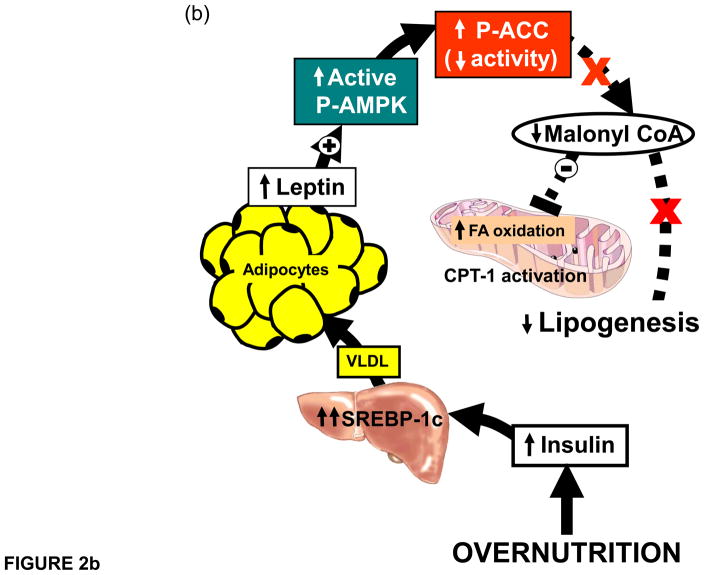
(a) Proposed pathway depicting the pathway to the metabolic syndrome. Chronic caloric surplus is here promoted to be the sine qua non for all subsequent events. It differs from more conventional views of the etiology of this disorder in three main respects: 1) Obesity, commonly considered a “disease” that causes metabolic syndrome, is depicted here as a normal physiologic response to caloric surplus that actually protects, at least temporarily, by sequestering toxic fatty acids in adipocytes that would otherwise damage organs. 2) Insulin resistance is also taken out of the etiologic mainstream to become a consequence, rather than cause, of the ectopic lipid deposition. 3) Leptin resistance is placed in a key causal role to explain why the hyperleptinemia of chronic overnutrition ultimately loses its ability to prevent ectopic lipid accumulation in target organs, at which point the metabolic syndrome is present. (b) Role of obesity-induced hyperleptinemia. Hyperleptinemia lowers fat content in peripheral organs. As adipocytes expand with triglycerides, leptin secretion increases proportionately. Since leptin stimulates fatty acid oxidation, adipocytes would be oxidizing, rather than storing, fat if the endogenous leptin they secrete were to act on them. Such an autocrine/paracrine relationship between the secretory product, leptin, and the secreting cells, is prevented by a progressive decline of leptin receptor expression. This physiologic leptin resistance is essential to permit accumulation of surplus calories into adipocytes. Meanwhile, the lipo-oxidative action of leptin is fully operative on peripheral organs, which minimizes ectopic lipid accumulation, at least temporarily. However, later in life peripheral organs also become leptin resistant. Leptin action on the hypothalamus limits the level of overnutrition without inhibiting it, while leptin’s lipooxidative action on the peripheral tissues keeps them free of ectopic lipid accumulation resulting from adipocyte spillover. If the disappearance of the leptin receptor during overnutrition is prevented by transgenic overexpression, obesity is prevented.
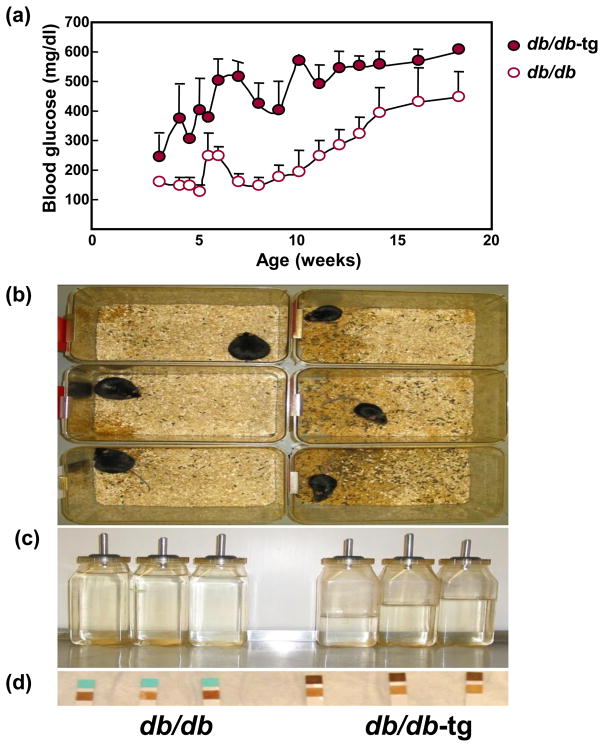
Comparison of (a) blood glucose levels, (b) wet sawdust, a reflection of polyuria, (c) water bottle volume, an index of polydipsia, and (d) glycosuria in obese db/db mice and age-matched db/db mice in which obesity was prevented by transgenic expression of the leptin receptor on an aP2 promoter (db/db-tg). Clearly the absence of obesity accelerates the appearance of T2DM by about 5 weeks. The transgene on adipocytes prevents adipocyte hypertrophy, which attenuates the usual obesity-associated hyperleptinemia. The limited adipogenesis in these mice was associated with marked increase in ectopic lipid deposition, again absolving obesity as a cause of metabolic syndrome.
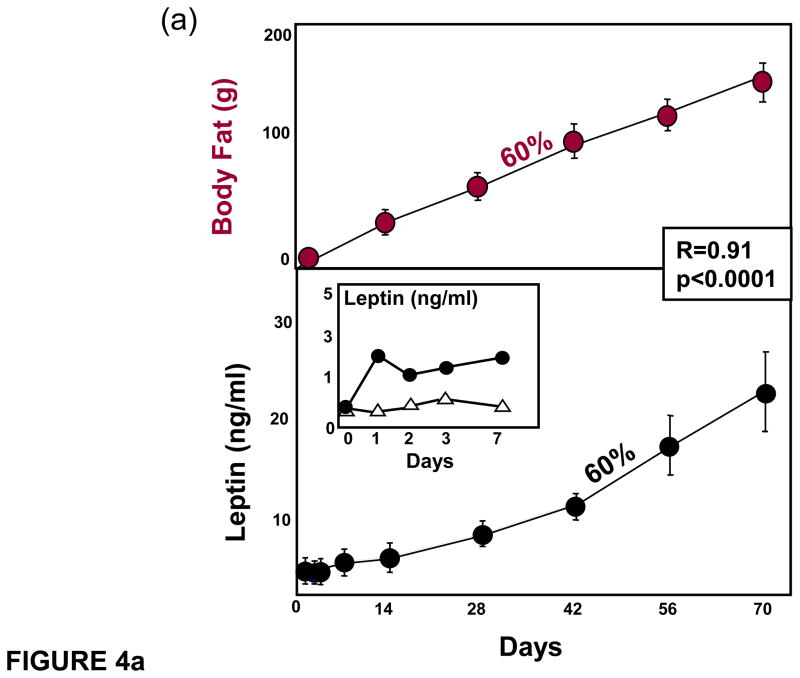
(a) Body fat and leptin mRNA of epididymal fat tissue in normal rats during the feeding of a 60% fat diet. Note their parallels. The rise in leptin begins within 1 day of high fat feeding (inset). (b) Comparison of body fat distribution in the normal rats fed a 60% diet (from panel a) versus unleptinized Zucker diabetic fatty (ZDF) rats fed only a 6% fat diet after 70 days.
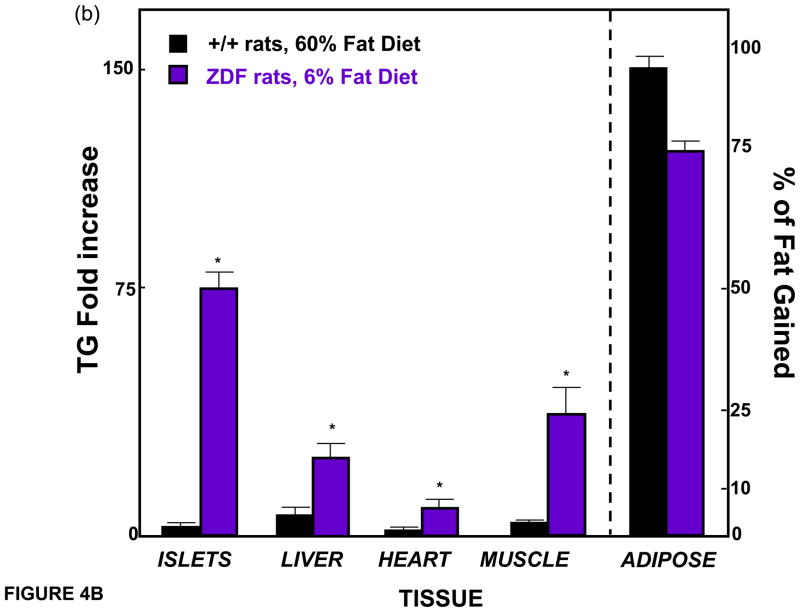
(a) Body fat and leptin mRNA of epididymal fat tissue in normal rats during the feeding of a 60% fat diet. Note their parallels. The rise in leptin begins within 1 day of high fat feeding (inset). (b) Comparison of body fat distribution in the normal rats fed a 60% diet (from panel a) versus unleptinized Zucker diabetic fatty (ZDF) rats fed only a 6% fat diet after 70 days.
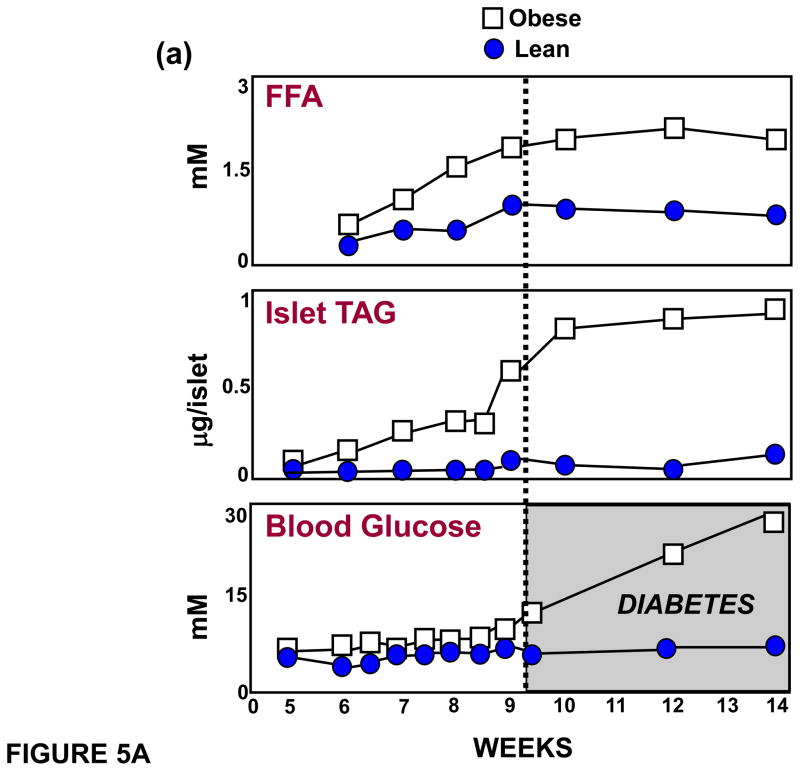
(a) Relationship between plasma free fatty acids (FFA), islet triglyceride content (TG) and blood glucose levels, demonstrating that the lipid abnormalities precede the onset of diabetes. (b) Evidence that a 1mM increase in FFA can induce apoptosis manifested by increased % of DNA laddering. This is blocked by 2mM L-cycloserine, which competitively inhibits ceramide formation that results from condensation of palmitoyl CoA and L-serine, catalyzed by serine palmitoyl transferase (SPT), the rate-limiting enzyme of de novo ceramide synthesis. (c) SPT mRNA is increased in islets of Zucker diabetic fatty (fa/fa) rat lacking leptin action. (d) SPT activity, assayed by measuring [3H]-palmitate incorporation into [3H]-ceramide, is also increased in unleptinized ZDF (fa/fa) islets.
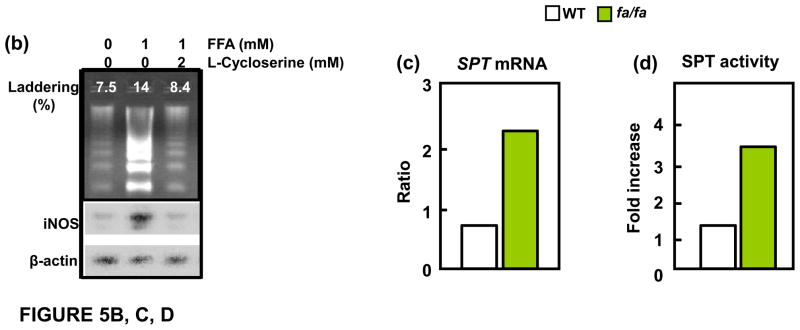
(a) Relationship between plasma free fatty acids (FFA), islet triglyceride content (TG) and blood glucose levels, demonstrating that the lipid abnormalities precede the onset of diabetes. (b) Evidence that a 1mM increase in FFA can induce apoptosis manifested by increased % of DNA laddering. This is blocked by 2mM L-cycloserine, which competitively inhibits ceramide formation that results from condensation of palmitoyl CoA and L-serine, catalyzed by serine palmitoyl transferase (SPT), the rate-limiting enzyme of de novo ceramide synthesis. (c) SPT mRNA is increased in islets of Zucker diabetic fatty (fa/fa) rat lacking leptin action. (d) SPT activity, assayed by measuring [3H]-palmitate incorporation into [3H]-ceramide, is also increased in unleptinized ZDF (fa/fa) islets.
References
-
- Cohen J, et al. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet. 2005;37:161–165. - PubMed
-
- Keys A. Human atherosclerosis and the diet. Circulation. 1952;5:115–118. - PubMed
-
- Unger RH. Lipotoxic diseases. Annu Rev Med. 2002;53:319–336. - PubMed
-
- Unger RH. Lipid overload and overflow: metabolic trauma and the metabolic syndrome. Trends Endocrinol Metab. 2003;14:398–403. - PubMed
-
- Unger RH. The physiology of cellular liporegulation. Annu Rev Physiol. 2003;65:333–347. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
