

How I use hydroxyurea to treat young patients with sickle cell anemia
- PMID: 20223921
- PMCID: PMC2902131
- DOI: 10.1182/blood-2009-04-146852
How I use hydroxyurea to treat young patients with sickle cell anemia
Abstract
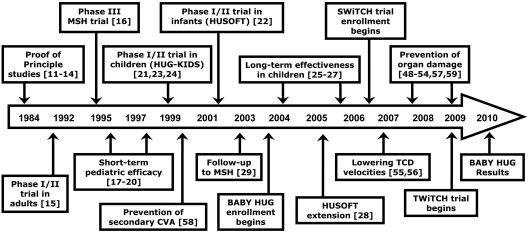
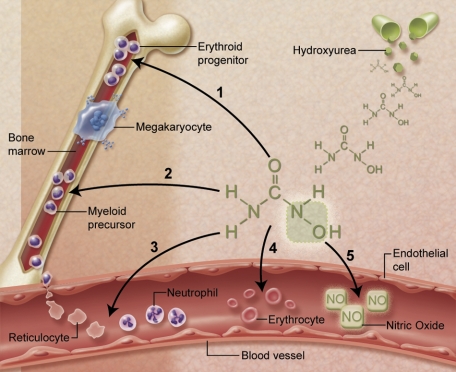
Hydroxyurea has many characteristics of an ideal drug for sickle cell anemia (SCA) and provides therapeutic benefit through multiple mechanisms of action. Over the past 25 years, substantial experience has accumulated regarding its safety and efficacy for patients with SCA. Early proof-of-principle studies were followed by prospective phase 1/2 trials demonstrating efficacy in affected adults, then adolescents and children, and more recently infants and toddlers. The phase 3 National Heart, Lung and Blood Institute-sponsored Multicenter Study of Hydroxyurea trial proved clinical efficacy for preventing acute vaso-occlusive events in severely affected adults. Based on this cumulative experience, hydroxyurea has emerged as an important therapeutic option for children and adolescents with recurrent vaso-occlusive events; recent evidence documents sustained long-term benefits with prevention or reversal of chronic organ damage. Despite abundant evidence for its efficacy, however, hydroxyurea has not yet translated into effective therapy for SCA. Because many healthcare providers have inadequate knowledge about hydroxyurea, patients and families are not offered treatment or decline because of unrealistic fears. Limited support for hydroxyurea by lay organizations and inconsistent medical delivery systems also contribute to underuse. Although questions remain regarding its long-term risks and benefits, current evidence suggests that many young patients with SCA should receive hydroxyurea treatment.
Figures
References
-
- Leikin SL, Gallagher D, Kinney TR, et al. Mortality in children and adolescents with sickle cell disease: Cooperative Study of Sickle Cell Disease. Pediatrics. 1989;84(3):500–508. - PubMed
-
- Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease: life expectancy and risk factors for early death. N Engl J Med. 1994;330(23):1639–1644. - PubMed
-
- Trompeter S, Roberts I. Haemoglobin F modulation in childhood sickle cell disease. Br J Haematol. 2009;144(3):308–316. - PubMed
-
- Ohene-Frempong K, Weiner SJ, Sleeper LA, et al. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood. 1998;91(1):288–294. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
