

Nrf2-regulated PPAR{gamma} expression is critical to protection against acute lung injury in mice
- PMID: 20224069
- PMCID: PMC2913232
- DOI: 10.1164/rccm.200907-1047OC
Nrf2-regulated PPAR{gamma} expression is critical to protection against acute lung injury in mice
Abstract
Rationale: The NF-E2 related factor 2 (Nrf2)-antioxidant response element (ARE) pathway is essential for protection against oxidative injury and inflammation including hyperoxia-induced acute lung injury. Microarray expression profiling revealed that lung peroxisome proliferator activated receptor gamma (PPARgamma) induction is suppressed in hyperoxia-susceptible Nrf2-deficient (Nrf2(-/-)) mice compared with wild-type (Nrf2(+/+)) mice. PPARgamma has pleiotropic beneficial effects including antiinflammation in multiple tissues.
Objectives: We tested the hypothesis that PPARgamma is an important determinant of pulmonary responsivity to hyperoxia regulated by Nrf2.
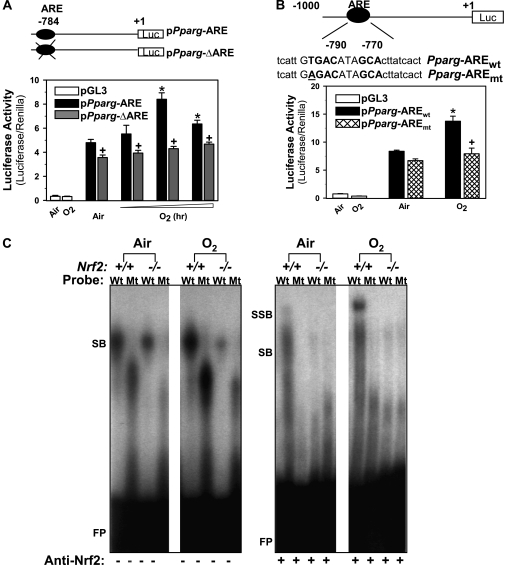
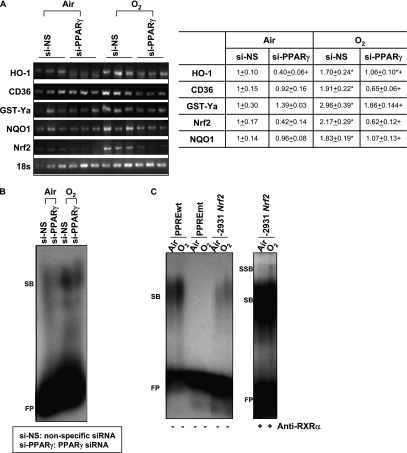
Methods: A computational bioinformatic method was applied to screen potential AREs in the Pparg promoter for Nrf2 binding. The functional role of a potential ARE was investigated by in vitro promoter analysis. A role for PPARgamma in hyperoxia-induced acute lung injury was determined by temporal silencing of PPARgamma via intranasal delivery of PPARgamma-specific interference RNA and by administration of a PPARgamma ligand 15-deoxy-Delta(12,14)-prostaglandin J(2) in mice.
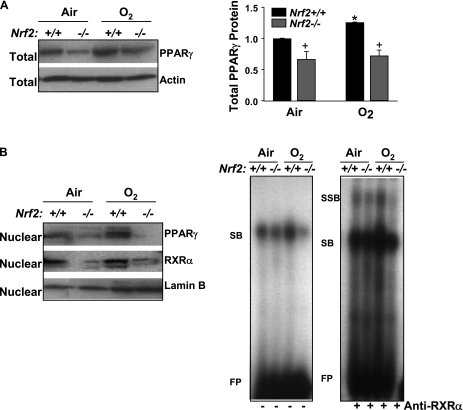
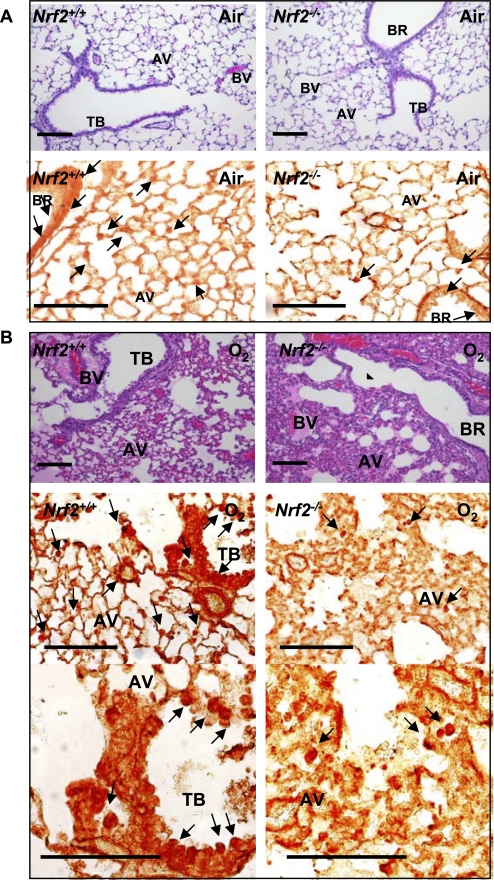
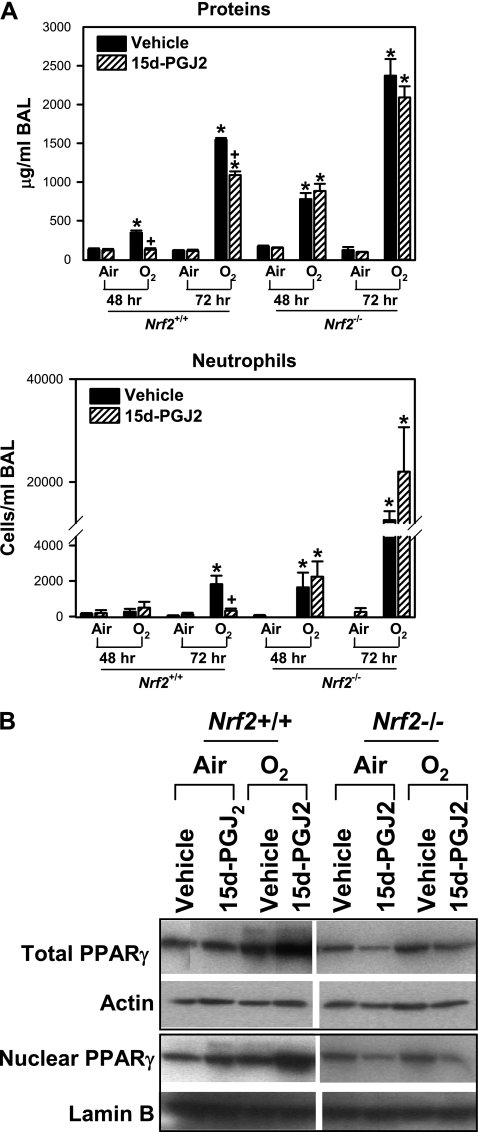
Measurements and main results: Deletion or site-directed mutagenesis of a potential ARE spanning -784/-764 sequence significantly attenuated hyperoxia-increased Pparg promoter activity in airway epithelial cells overexpressing Nrf2, indicating that the -784/-764 ARE is critical for Nrf2-regulated PPARgamma expression. Mice with decreased lung PPARgamma by specific interference RNA treatment had significantly augmented hyperoxia-induced pulmonary inflammation and injury. 15 Deoxy-Delta(12,14)-prostaglandin J(2) administration significantly reduced hyperoxia-induced lung inflammation and edema in Nrf2(+/+), but not in Nrf2(-/-) mice.
Conclusions: Results indicate for the first time that Nrf2-driven PPARgamma induction has an essential protective role in pulmonary oxidant injury. Our observations provide new insights into the therapeutic potential of PPARgamma in airway oxidative inflammatory disorders.
Figures



Comment in
-
Nrf2 and PPAR{gamma}: PPARtnering against oxidant-induced lung injury.Am J Respir Crit Care Med. 2010 Jul 15;182(2):134-5. doi: 10.1164/rccm.201004-0457ED. Am J Respir Crit Care Med. 2010. PMID: 20634499 No abstract available.
Similar articles
-
Nrf2 and PPAR{gamma}: PPARtnering against oxidant-induced lung injury.Am J Respir Crit Care Med. 2010 Jul 15;182(2):134-5. doi: 10.1164/rccm.201004-0457ED. Am J Respir Crit Care Med. 2010. PMID: 20634499 No abstract available.
-
Sulforaphane enriched transcriptome of lung mitochondrial energy metabolism and provided pulmonary injury protection via Nrf2 in mice.Toxicol Appl Pharmacol. 2019 Feb 1;364:29-44. doi: 10.1016/j.taap.2018.12.004. Epub 2018 Dec 5. Toxicol Appl Pharmacol. 2019. PMID: 30529165 Free PMC article.
-
Transactivation of the PPAR-responsive enhancer module in chemopreventive glutathione S-transferase gene by the peroxisome proliferator-activated receptor-gamma and retinoid X receptor heterodimer.Cancer Res. 2004 May 15;64(10):3701-13. doi: 10.1158/0008-5472.CAN-03-3924. Cancer Res. 2004. PMID: 15150131
-
Nrf2 protects against airway disorders.Toxicol Appl Pharmacol. 2010 Apr 1;244(1):43-56. doi: 10.1016/j.taap.2009.07.024. Epub 2009 Jul 29. Toxicol Appl Pharmacol. 2010. PMID: 19646463 Review.
-
Collaborative Power of Nrf2 and PPARγ Activators against Metabolic and Drug-Induced Oxidative Injury.Oxid Med Cell Longev. 2017;2017:1378175. doi: 10.1155/2017/1378175. Epub 2017 Aug 27. Oxid Med Cell Longev. 2017. PMID: 28928902 Free PMC article. Review.
Cited by
-
[Research Progress of Nrf2 and Ferroptosis in Tumor Drug Resistance].Zhongguo Fei Ai Za Zhi. 2023 Oct 20;26(10):765-773. doi: 10.3779/j.issn.1009-3419.2023.101.31. Zhongguo Fei Ai Za Zhi. 2023. PMID: 37989339 Free PMC article. Review. Chinese.
-
NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis.Redox Biol. 2019 May;23:101107. doi: 10.1016/j.redox.2019.101107. Epub 2019 Jan 11. Redox Biol. 2019. PMID: 30692038 Free PMC article. Review.
-
The Nrf2/ARE Pathway: A Promising Target to Counteract Mitochondrial Dysfunction in Parkinson's Disease.Parkinsons Dis. 2011 Feb 22;2011:314082. doi: 10.4061/2011/314082. Parkinsons Dis. 2011. PMID: 21403858 Free PMC article.
-
Emerging perspectives of copper-mediated transcriptional regulation in mammalian cell development.Metallomics. 2024 Oct 4;16(10):mfae046. doi: 10.1093/mtomcs/mfae046. Metallomics. 2024. PMID: 39375833 Free PMC article. Review.
-
Electroacupuncture Pretreatment Alleviates LPS-Induced Acute Respiratory Distress Syndrome via Regulating the PPAR Gamma/NF-Kappa B Signaling Pathway.Evid Based Complement Alternat Med. 2020 Jul 22;2020:4594631. doi: 10.1155/2020/4594631. eCollection 2020. Evid Based Complement Alternat Med. 2020. PMID: 32774418 Free PMC article.
References
-
- Bowler RP, Barnes PJ, Crapo JD. The role of oxidative stress in chronic obstructive pulmonary disease. COPD 2004;1:255–277. - PubMed
-
- Chow CW, Herrera Abreu MT, Suzuki T, Downey GP. Oxidative stress and acute lung injury. Am J Respir Cell Mol Biol 2003;29:427–431. - PubMed
-
- Paz-Elizur T, Krupsky M, Elinger D, Schechtman E, Livneh Z. Repair of the oxidative DNA damage 8-oxoguanine as a biomarker for lung cancer risk. Cancer Biomark 2005;1:201–205. - PubMed
-
- Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med 2000;342:1334–1349. - PubMed
-
- Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, et al. An Nrf2/small MAF heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun 1997;236:313–322. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
