

FastTree 2--approximately maximum-likelihood trees for large alignments
- PMID: 20224823
- PMCID: PMC2835736
- DOI: 10.1371/journal.pone.0009490
FastTree 2--approximately maximum-likelihood trees for large alignments
Abstract
Background: We recently described FastTree, a tool for inferring phylogenies for alignments with up to hundreds of thousands of sequences. Here, we describe improvements to FastTree that improve its accuracy without sacrificing scalability.
Methodology/principal findings: Where FastTree 1 used nearest-neighbor interchanges (NNIs) and the minimum-evolution criterion to improve the tree, FastTree 2 adds minimum-evolution subtree-pruning-regrafting (SPRs) and maximum-likelihood NNIs. FastTree 2 uses heuristics to restrict the search for better trees and estimates a rate of evolution for each site (the "CAT" approximation). Nevertheless, for both simulated and genuine alignments, FastTree 2 is slightly more accurate than a standard implementation of maximum-likelihood NNIs (PhyML 3 with default settings). Although FastTree 2 is not quite as accurate as methods that use maximum-likelihood SPRs, most of the splits that disagree are poorly supported, and for large alignments, FastTree 2 is 100-1,000 times faster. FastTree 2 inferred a topology and likelihood-based local support values for 237,882 distinct 16S ribosomal RNAs on a desktop computer in 22 hours and 5.8 gigabytes of memory.
Conclusions/significance: FastTree 2 allows the inference of maximum-likelihood phylogenies for huge alignments. FastTree 2 is freely available at http://www.microbesonline.org/fasttree.
Conflict of interest statement
Figures
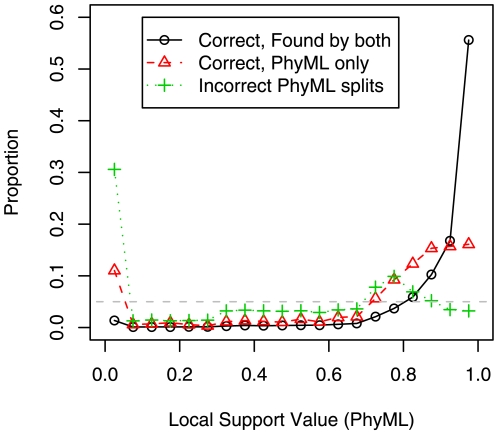
 + SPRs on simulated alignments with 250 protein sequences. We classified PhyML's splits as correct and found by both PhyML and FastTree, correct but missed by FastTree, or incorrect. We show the distribution of support values for each class. The right-most bin includes the strongly supported splits (0.95 to 1.0), and the gray dashed line shows the uniform distribution. The support values are PhyML's minimum of the approximate likelihood ratio test and SH-like , local supports.
+ SPRs on simulated alignments with 250 protein sequences. We classified PhyML's splits as correct and found by both PhyML and FastTree, correct but missed by FastTree, or incorrect. We show the distribution of support values for each class. The right-most bin includes the strongly supported splits (0.95 to 1.0), and the gray dashed line shows the uniform distribution. The support values are PhyML's minimum of the approximate likelihood ratio test and SH-like , local supports.
 axis). For RAxML with FastTree's (minimum-evolution) starting tree, we show the starting topology and RAxML's first two rounds of SPR moves.
axis). For RAxML with FastTree's (minimum-evolution) starting tree, we show the starting topology and RAxML's first two rounds of SPR moves.
References
-
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. - PubMed
-
- Studier JA, Keppler KJ. A note on the neighbor-joining algorithm of Saitou and Nei. Mol Biol Evol. 1988;5:729–31. - PubMed
-
- Felsenstein J. Evolutionary trees from dna sequences: A maximum likelihood approach. J Mol Evol. 1981;17:368–376. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
