

Pathogenesis and treatment of mitochondrial disorders
- PMID: 20225024
- PMCID: PMC10440730
- DOI: 10.1007/978-90-481-2813-6_10
Pathogenesis and treatment of mitochondrial disorders
Abstract
In the past 50 years, our understanding of the biochemical and molecular causes of mitochondrial diseases, defined restrictively as disorders due to defects of the mitochondrial respiratory chain (RC), has made great strides. Mitochondrial diseases can be due to mutations in mitochondrial DNA (mtDNA) or in nuclear DNA (nDNA) and each group can be subdivided into more specific classes. Thus, mtDNA-related disorders can result from mutations in genes affecting protein synthesis in toto or mutations in protein-coding genes. Mendelian mitochondrial disorders can be attributed to mutations in genes that (i) encode subunits of the RC ("direct hits"); (ii) encode assembly proteins or RC complexes ("indirect hits"); (iii) encode factors needed for mtDNA maintenance, replication, or translation (intergenomic signaling); (iv) encode components of the mitochondrial protein import machinery; (v) control the synthesis and composition of mitochondrial membrane phospholipids; and (vi) encode proteins involved in mitochondrial dynamics.In contrast to this wealth of knowledge about etiology, our understanding of pathogenic mechanism is very limited. We discuss pathogenic factors that can influence clinical expression, especially ATP shortage and reactive oxygen radicals (ROS) excess. Therapeutic options are limited and fall into three modalities: (i) symptomatic interventions, which are palliative but crucial for day-to-day management; (ii) radical approaches aimed at correcting the biochemical or molecular error, which are interesting but still largely experimental; and (iii) pharmacological means of interfering with the pathogenic cascade of events (e.g. boosting ATP production or scavenging ROS), which are inconsistently and incompletely effective, but can be safe and helpful.
Figures
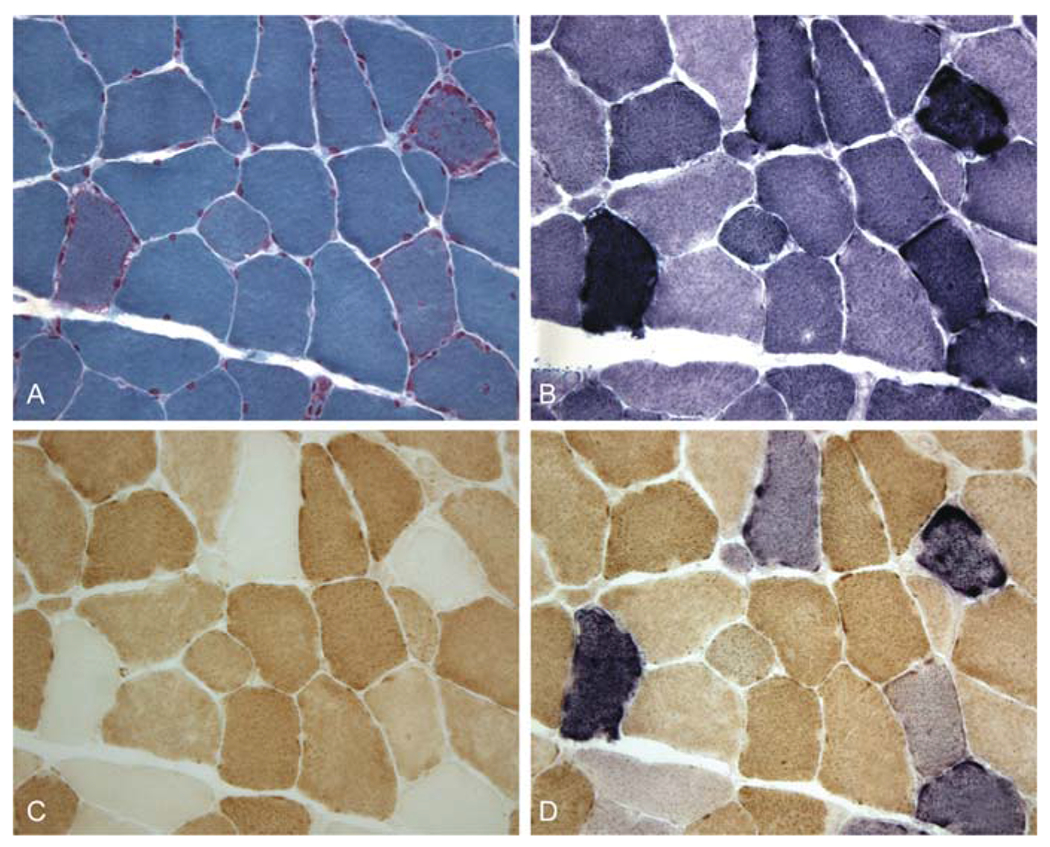
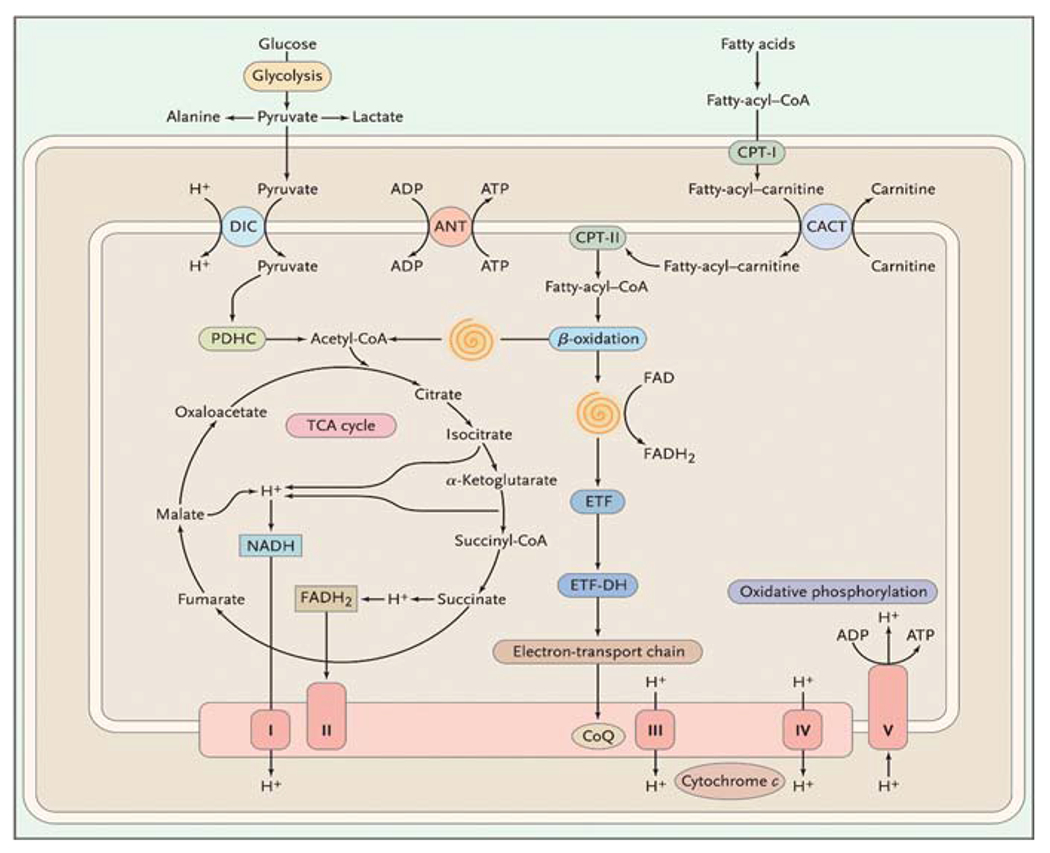
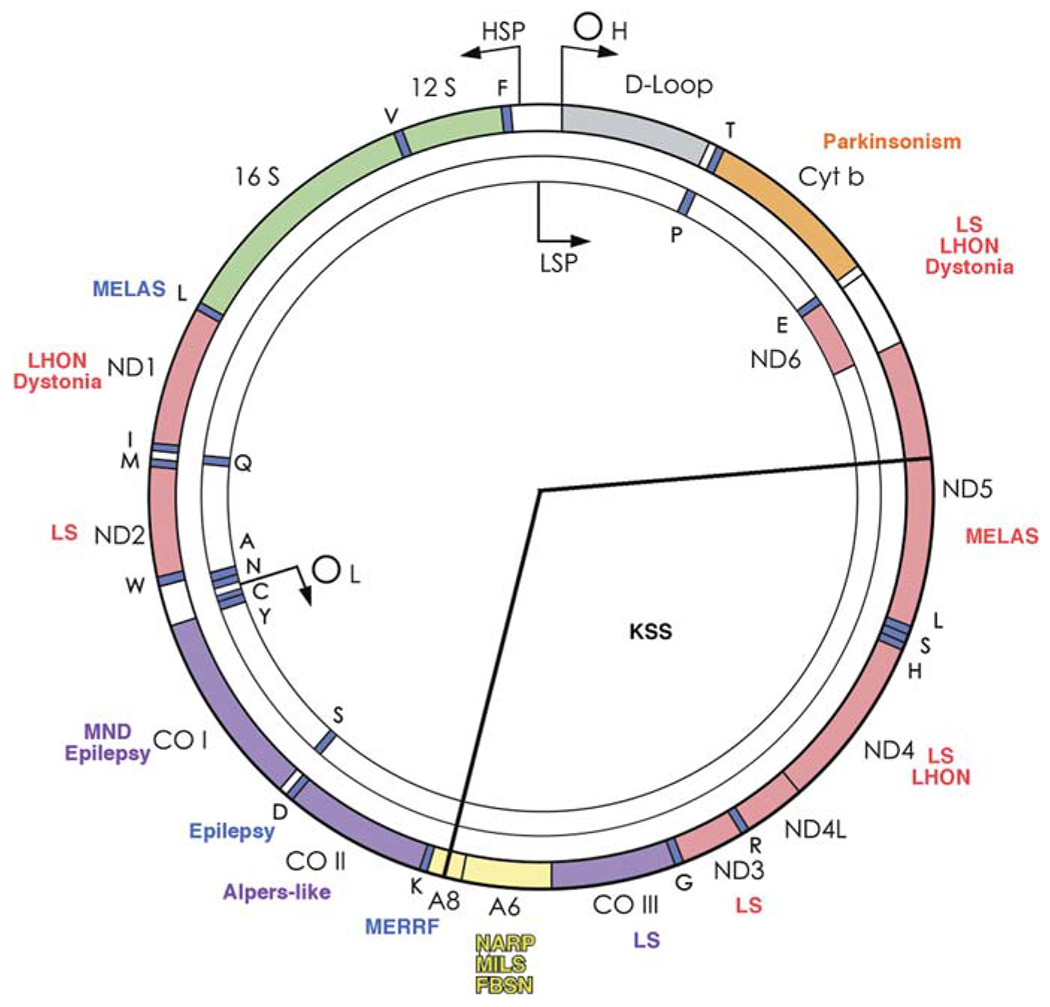
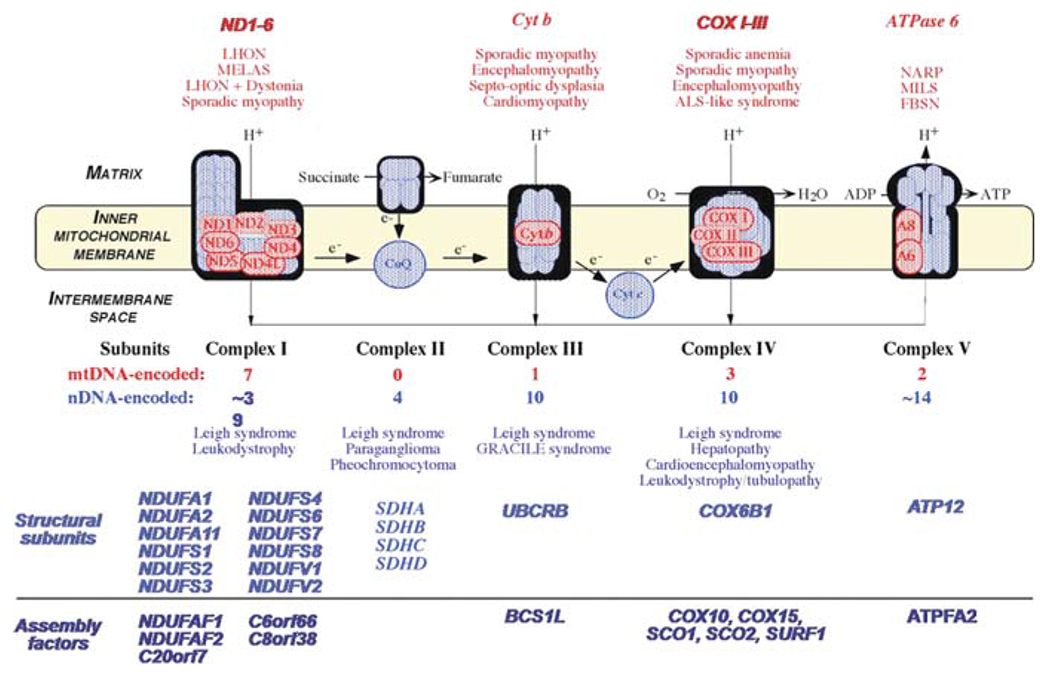
Similar articles
-
Mitochondrial diseases.Biochim Biophys Acta. 2004 Jul 23;1658(1-2):80-8. doi: 10.1016/j.bbabio.2004.03.014. Biochim Biophys Acta. 2004. PMID: 15282178 Review.
-
Mitochondrial encephalomyopathies: an update.Neuromuscul Disord. 2005 Apr;15(4):276-86. doi: 10.1016/j.nmd.2004.12.008. Neuromuscul Disord. 2005. PMID: 15792866 Review.
-
A history of mitochondrial diseases.J Inherit Metab Dis. 2011 Apr;34(2):261-76. doi: 10.1007/s10545-010-9082-x. Epub 2010 May 21. J Inherit Metab Dis. 2011. PMID: 20490929
-
Infantile-onset disorders of mitochondrial replication and protein synthesis.J Child Neurol. 2011 Jul;26(7):866-75. doi: 10.1177/0883073811402072. Epub 2011 May 13. J Child Neurol. 2011. PMID: 21572058 Review.
-
Pathogenic mutations of nuclear genes associated with mitochondrial disorders.Acta Biochim Biophys Sin (Shanghai). 2009 Mar;41(3):179-87. doi: 10.1093/abbs/gmn021. Acta Biochim Biophys Sin (Shanghai). 2009. PMID: 19280056 Review.
Cited by
-
Mitochondrial Dysfunction and Cardiovascular Disease: Pathophysiology and Emerging Therapies.Oxid Med Cell Longev. 2022 Aug 2;2022:9530007. doi: 10.1155/2022/9530007. eCollection 2022. Oxid Med Cell Longev. 2022. Retraction in: Oxid Med Cell Longev. 2024 Jan 9;2024:9785792. doi: 10.1155/2024/9785792. PMID: 35958017 Free PMC article. Retracted. Review.
-
Mitochondria in the maintenance of hematopoietic stem cells: new perspectives and opportunities.Blood. 2019 May 2;133(18):1943-1952. doi: 10.1182/blood-2018-10-808873. Epub 2019 Feb 26. Blood. 2019. PMID: 30808633 Free PMC article. Review.
-
The Impact of Heritable Myopathies on Gastrointestinal Skeletal Muscle Function.Cell Mol Gastroenterol Hepatol. 2025;19(8):101522. doi: 10.1016/j.jcmgh.2025.101522. Epub 2025 Apr 22. Cell Mol Gastroenterol Hepatol. 2025. PMID: 40268053 Free PMC article. Review.
-
Mitochondrial Dynamics Regulation in Skin Fibroblasts from Mitochondrial Disease Patients.Biomolecules. 2020 Mar 13;10(3):0. doi: 10.3390/biom10030450. Biomolecules. 2020. PMID: 32183225 Free PMC article.
-
Cellular stress responses and dysfunctional Mitochondrial-cellular senescence, and therapeutics in chronic respiratory diseases.Redox Biol. 2020 Jun;33:101443. doi: 10.1016/j.redox.2020.101443. Epub 2020 Jan 25. Redox Biol. 2020. PMID: 32037306 Free PMC article. Review.
References
-
- Wallace DC, Singh G, Lott MT, et al. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 1988;242:1427–1430. - PubMed
-
- Holt IJ, Harding AE, Morgan Hughes JA. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 1988;331:717–719. - PubMed
-
- Gvozdjáková A, ed. Mitochondrial Medicine: Springer, 2008.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
