

Dynamics of gene-modified progenitor cells analyzed by tracking retroviral integration sites in a human SCID-X1 gene therapy trial
- PMID: 20228274
- PMCID: PMC2881494
- DOI: 10.1182/blood-2009-12-257352
Dynamics of gene-modified progenitor cells analyzed by tracking retroviral integration sites in a human SCID-X1 gene therapy trial
Abstract
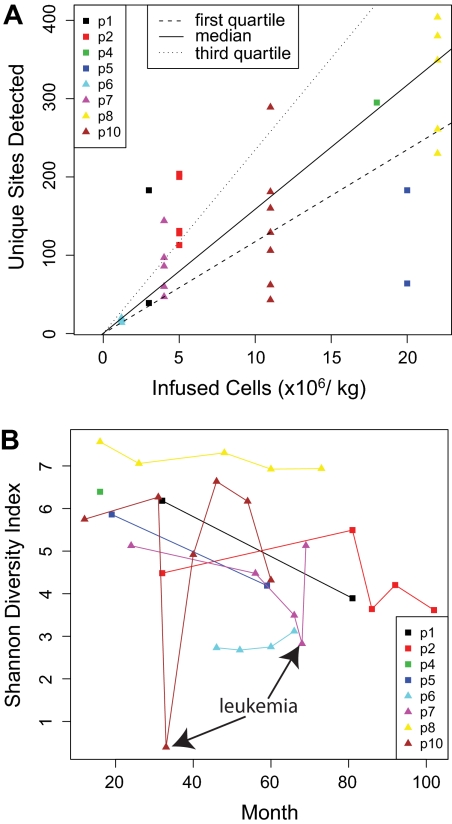
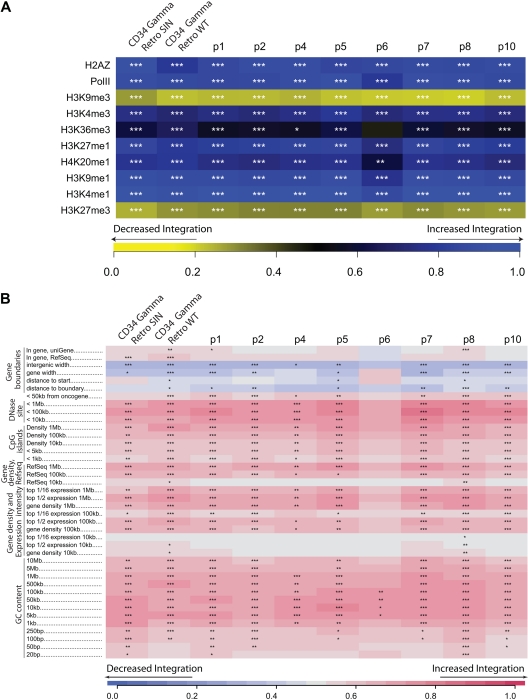
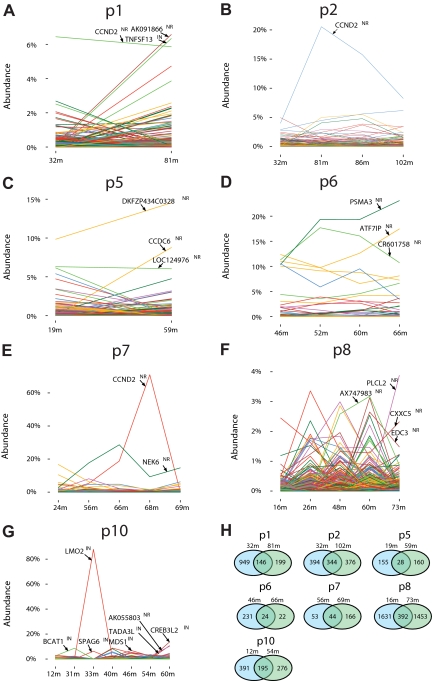
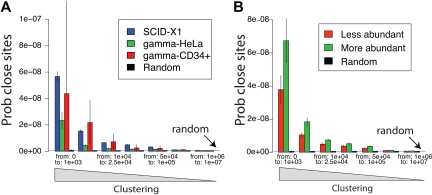
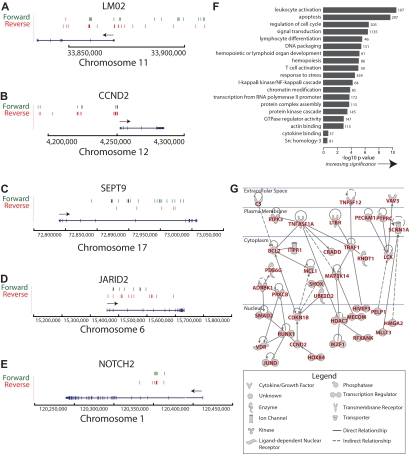
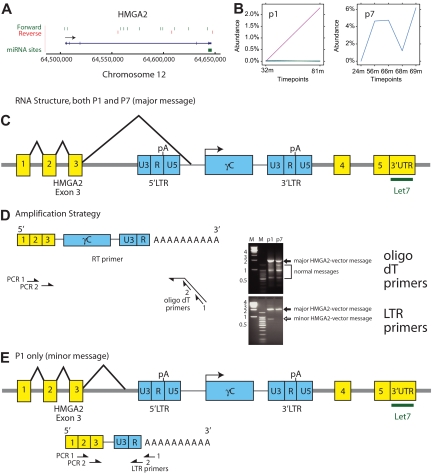
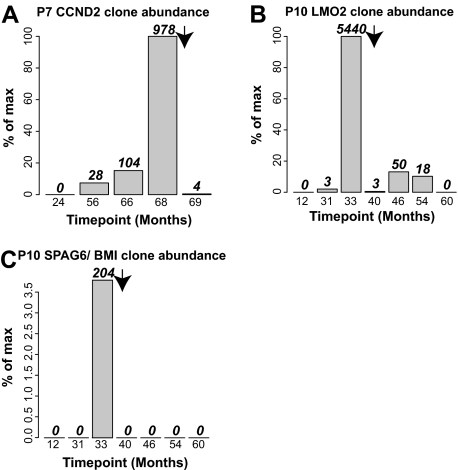
X-linked severe-combined immunodeficiency (SCID-X1) has been treated by therapeutic gene transfer using gammaretroviral vectors, but insertional activation of proto-oncogenes contributed to leukemia in some patients. Here we report a longitudinal study of gene-corrected progenitor cell populations from 8 patients using 454 pyrosequencing to map vector integration sites, and extensive resampling to allow quantification of clonal abundance. The number of transduced cells infused into patients initially predicted the subsequent diversity of circulating cells. A capture-recapture analysis was used to estimate the size of the gene-corrected cell pool, revealing that less than 1/100th of the infused cells had long-term repopulating activity. Integration sites were clustered even at early time points, often near genes involved in growth control, and several patients harbored expanded cell clones with vectors integrated near the cancer-implicated genes CCND2 and HMGA2, but remain healthy. Integration site tracking also documented that chemotherapy for adverse events resulted in successful control. The longitudinal analysis emphasizes that key features of transduced cell populations--including diversity, integration site clustering, and expansion of some clones--were established early after transplantation. The approaches to sequencing and bioinformatics analysis reported here should be widely useful in assessing the outcome of gene therapy trials.
Figures
References
-
- Cavazzana-Calvo M, Hacein-Bey S, de Saint Basile G, et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science. 2000;288(5466):669–672. - PubMed
-
- Hacein-Bey-Abina S, Le Deist F, Carlier F, et al. Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy. N Engl J Med. 2002;346(16):1185–1193. - PubMed
-
- Aiuti A, Slavin S, Aker M, et al. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science. 2002;296(5577):2410–2413. - PubMed
-
- Ott MG, Schmidt M, Schwarzwaelder K, et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat Med. 2006;12(4):401–409. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
