

Imatinib resistance associated with BCR-ABL upregulation is dependent on HIF-1alpha-induced metabolic reprograming
- PMID: 20228846
- PMCID: PMC2874611
- DOI: 10.1038/onc.2010.67
Imatinib resistance associated with BCR-ABL upregulation is dependent on HIF-1alpha-induced metabolic reprograming
Abstract
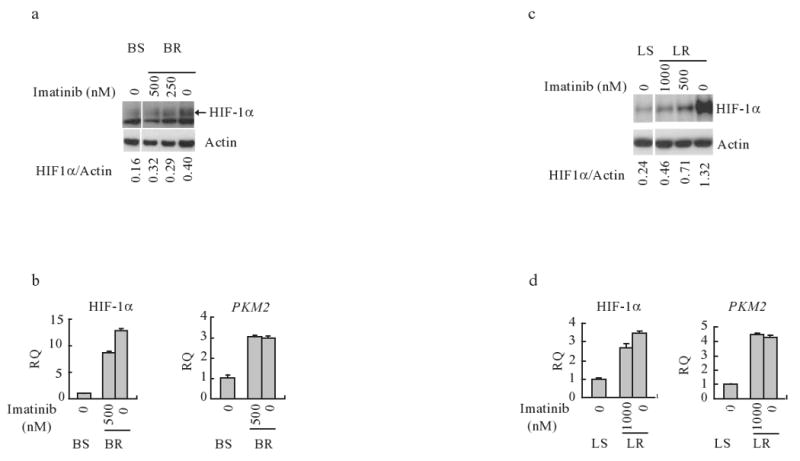
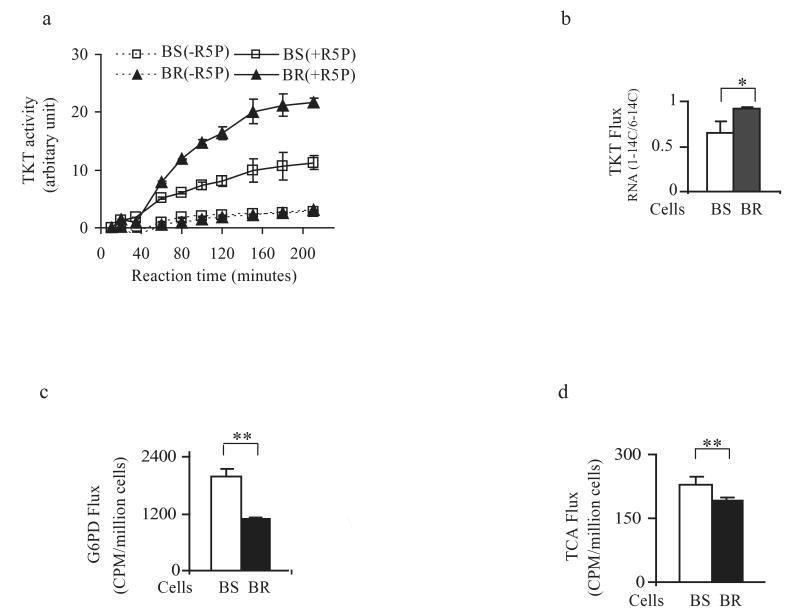
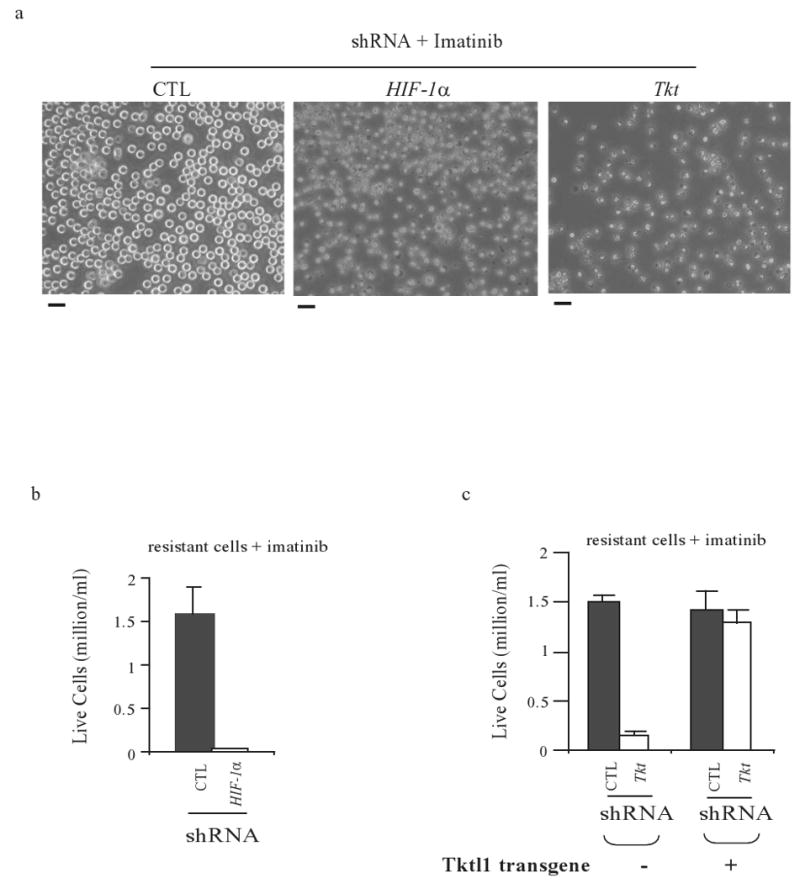
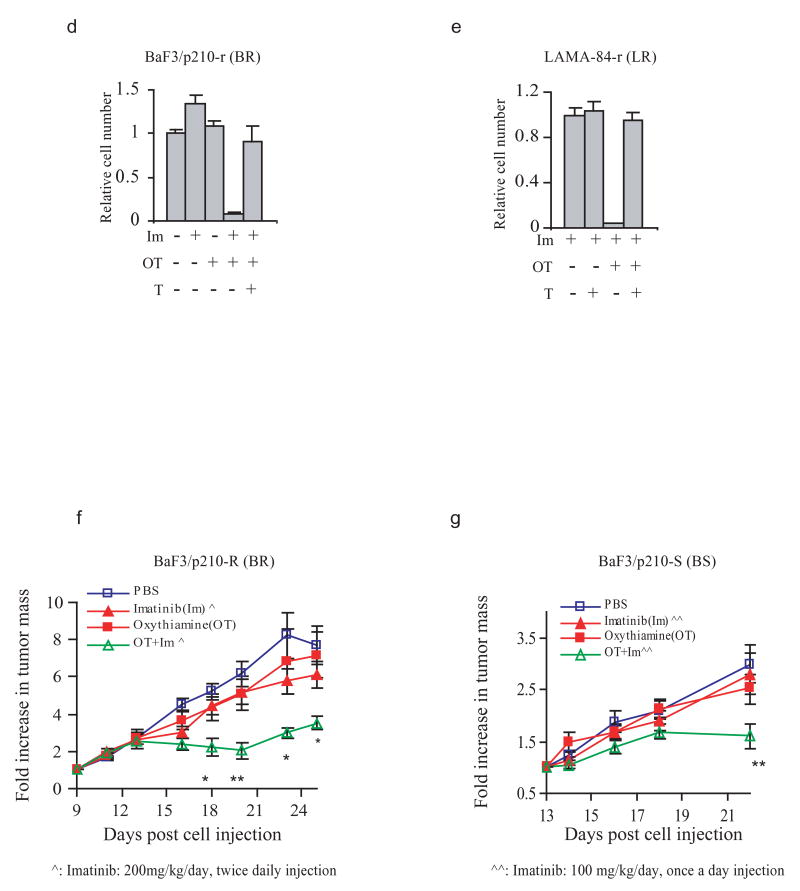
As chronic myeloid leukemia (CML) progresses from the chronic phase to blast crisis, the levels of BCR-ABL increase. In addition, blast-transformed leukemic cells display enhanced resistance to imatinib in the absence of BCR-ABL-resistance mutations. In this study, we show that when BCR-ABL-transformed cell lines were selected for imatinib resistance in vitro, the cells that grew out displayed a higher BCR-ABL expression comparable to the increase seen in accelerated forms of the disease. This enhanced expression of BCR-ABL was associated with an increased rate of glycolysis but with a decreased rate of proliferation. The higher level of BCR-ABL expression in the selected cells correlated with a nonhypoxic induction of hypoxia-inducible factor-1alpha (HIF-1alpha) that was required for cells to tolerate enhanced BCR-ABL signaling. HIF-1alpha induction resulted in an enhanced rate of glycolysis but with reduced glucose flux through both the tricarboxylic acid cycle and the oxidative arm of the pentose phosphate pathway (PPP). The reduction in oxidative PPP-mediated ribose synthesis was compensated by the HIF-1alpha-dependent activation of the nonoxidative PPP enzyme, transketolase, in imatinib-resistant CML cells. In both primary cultures of cells from patients exhibiting blast transformation and in vivo xenograft tumors, use of oxythiamine, which can inhibit both the pyruvate dehydrogenase complex and transketolase, resulted in enhanced imatinib sensitivity of tumor cells. Together, these results suggest that oxythiamine can enhance imatinib efficacy in patients who present an accelerated form of the disease.
Figures



References
-
- Barnes DJ, Palaiologou D, Panousopoulou E, Schultheis B, Yong AS, Wong A, et al. Bcr-Abl expression levels determine the rate of development of resistance to imatinib mesylate in chronic myeloid leukemia. Cancer Res. 2005a;65:8912–9. - PubMed
-
- Barnes K, McIntosh E, Whetton AD, Daley GQ, Bentley J, Baldwin SA. Chronic myeloid leukaemia: an investigation into the role of Bcr-Abl-induced abnormalities in glucose transport regulation. Oncogene. 2005b;24:3257–67. - PubMed
-
- Bell EL, Chandel NS. Mitochondrial oxygen sensing: regulation of hypoxia-inducible factor by mitochondrial generated reactive oxygen species. Essays Biochem. 2007;43:17–27. - PubMed
-
- Carroll M, Ohno-Jones S, Tamura S, Buchdunger E, Zimmermann J, Lydon NB, et al. CGP 57148, a tyrosine kinase inhibitor, inhibits the growth of cells expressing BCR-ABL, TEL-ABL, and TEL-PDGFR fusion proteins. Blood. 1997;90:4947–52. - PubMed
-
- Chamberlain BR, Buttery JE, Pannall PR. A stable reagent mixture for the whole blood transketolase assay. Ann Clin Biochem. 1996;33(Pt 4):352–4. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
