

Hepatitis C virus regulates transforming growth factor beta1 production through the generation of reactive oxygen species in a nuclear factor kappaB-dependent manner
- PMID: 20230822
- PMCID: PMC2883661
- DOI: 10.1053/j.gastro.2010.03.008
Hepatitis C virus regulates transforming growth factor beta1 production through the generation of reactive oxygen species in a nuclear factor kappaB-dependent manner
Abstract
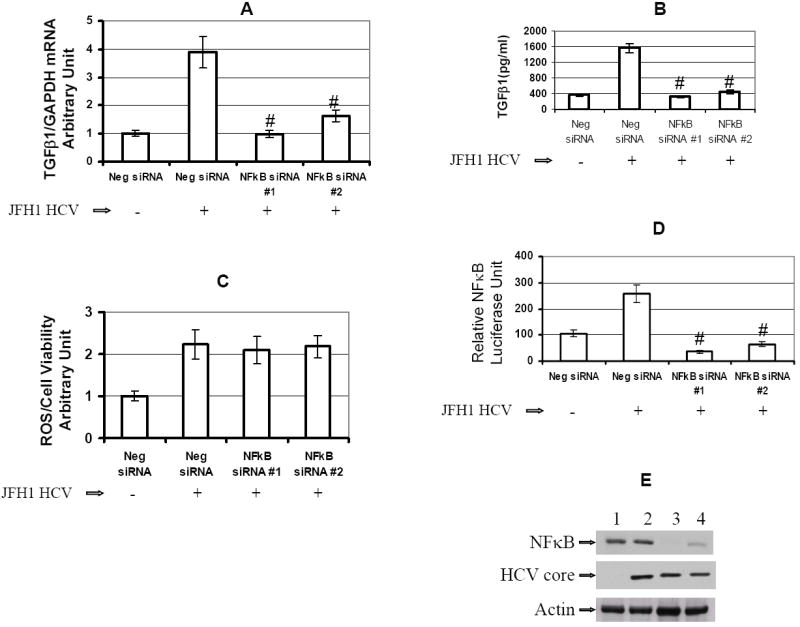
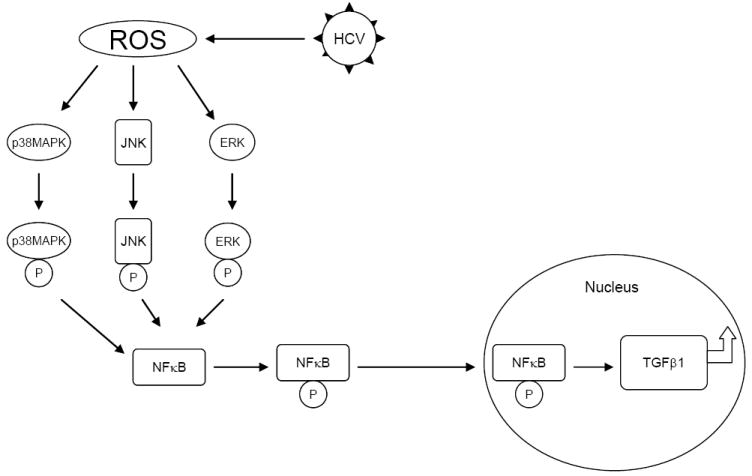
Background & aims: The generation of oxidative stress and transforming growth factor beta1 (TGF-beta1) production play important roles in liver fibrogenesis. We have previously shown that hepatitis C virus (HCV) increases hepatocyte TGF-beta1 expression. However, the mechanisms by which this induction occurs have not been well studied. We explored the possibility that HCV infection regulates TGF-beta1 expression through the generation of reactive oxygen species (ROS), which act through > or =1 of the p38 mitogen-activated protein kinase (MAPK), extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and nuclear factor kappaB (NFkappaB) signaling pathways to induce TGF-beta1 expression.
Methods: We used small molecule inhibitors and short interfering RNAs to knock down these pathways to study the mechanism by which HCV regulates TGF-beta1 production in the infectious JFH1 model.
Results: We demonstrated that HCV induces ROS and TGF-beta1 expression. We further found that JFH1 induces the phosphorylation of p38MAPK, JNK, ERK, and NFkappaB. We also found that HCV-mediated TGF-beta1 enhancement occurs through a ROS-induced and p38 MAPK, JNK, ERK1/2, NFkappaB-dependent pathway.
Conclusions: These findings provide further evidence to support the hypothesis that HCV enhances hepatic fibrosis progression through the generation of ROS and induction of TGF-beta1. Strategies to limit the viral induction of oxidative stress appear to be warranted to inhibit fibrogenesis.
Copyright 2010 AGA Institute. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
There is no conflict to disclose for the authors.
Figures





References
-
- Alter MJ. Epidemiology of viral hepatitis and HIV co-infection. J Hepatol. 2006;44:S6–9. - PubMed
-
- Lauer GM, Walker BD. Hepatitis C virus infection. N Engl J Med. 2001;345:41–52. - PubMed
-
- Bataller R, Paik YH, Lindquist JN, Lemasters JJ, Brenner DA. Hepatitis C virus core and nonstructural proteins induce fibrogenic effects in hepatic stellate cells. Gastroenterology. 2004;126:529–40. - PubMed
-
- Lin W, Weinberg EM, Tai AW, Peng LF, Brockman MA, Kim KA, Kim SS, Borges CB, Shao RX, Chung RT. HIV increases HCV replication in a TGF-beta1-dependent manner. Gastroenterology. 2008;134:803–11. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
