

Inhibition of acyl-coenzyme A:cholesterol acyltransferase 2 (ACAT2) prevents dietary cholesterol-associated steatosis by enhancing hepatic triglyceride mobilization
- PMID: 20231283
- PMCID: PMC2863169
- DOI: 10.1074/jbc.M110.118422
Inhibition of acyl-coenzyme A:cholesterol acyltransferase 2 (ACAT2) prevents dietary cholesterol-associated steatosis by enhancing hepatic triglyceride mobilization
Abstract
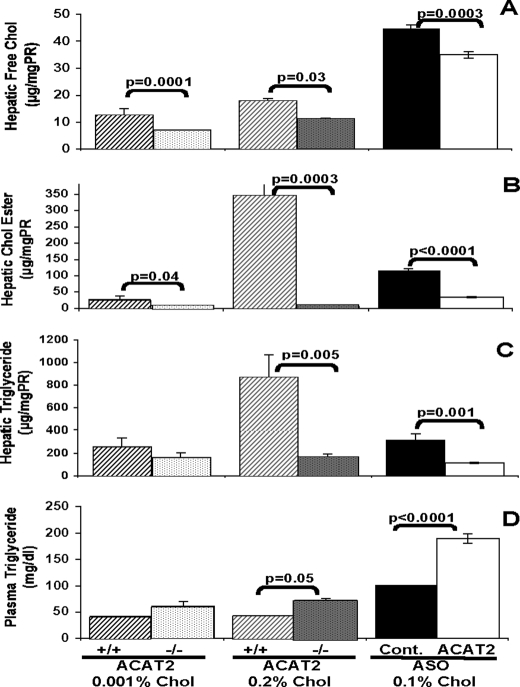
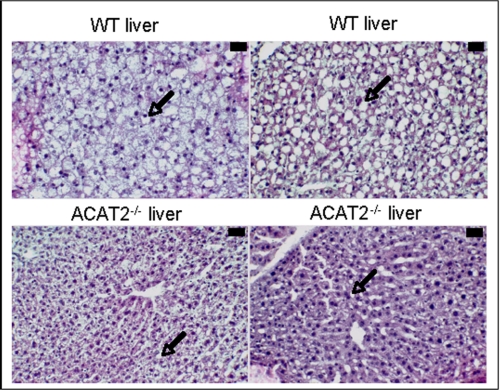
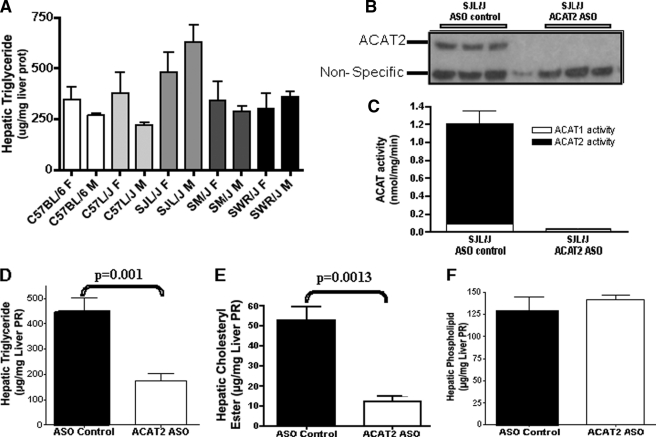
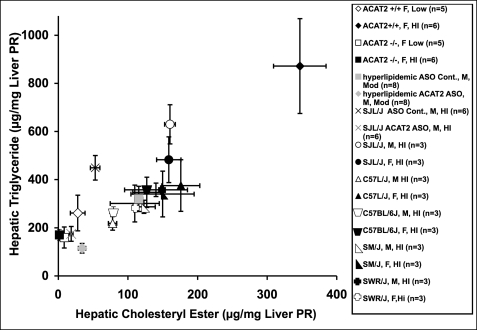
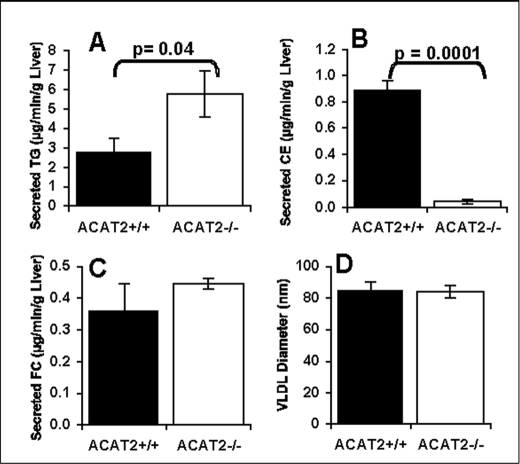
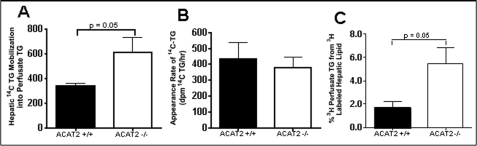
Acyl-CoA:cholesterol O-acyl transferase 2 (ACAT2) promotes cholesterol absorption by the intestine and the secretion of cholesteryl ester-enriched very low density lipoproteins by the liver. Paradoxically, mice lacking ACAT2 also exhibit mild hypertriglyceridemia. The present study addresses the unexpected role of ACAT2 in regulation of hepatic triglyceride (TG) metabolism. Mouse models of either complete genetic deficiency or pharmacological inhibition of ACAT2 were fed low fat diets containing various amounts of cholesterol to induce hepatic steatosis. Mice genetically lacking ACAT2 in both the intestine and the liver were dramatically protected against hepatic neutral lipid (TG and cholesteryl ester) accumulation, with the greatest differences occurring in situations where dietary cholesterol was elevated. Further studies demonstrated that liver-specific depletion of ACAT2 with antisense oligonucleotides prevents dietary cholesterol-associated hepatic steatosis both in an inbred mouse model of non-alcoholic fatty liver disease (SJL/J) and in a humanized hyperlipidemic mouse model (LDLr(-/-), apoB(100/100)). All mouse models of diminished ACAT2 function showed lowered hepatic triglyceride concentrations and higher plasma triglycerides secondary to increased hepatic secretion of TG into nascent very low density lipoproteins. This work demonstrates that inhibition of hepatic ACAT2 can prevent dietary cholesterol-driven hepatic steatosis in mice. These data provide the first evidence to suggest that ACAT2-specific inhibitors may hold unexpected therapeutic potential to treat both atherosclerosis and non-alcoholic fatty liver disease.
Figures
Similar articles
-
Liver-specific inhibition of acyl-coenzyme a:cholesterol acyltransferase 2 with antisense oligonucleotides limits atherosclerosis development in apolipoprotein B100-only low-density lipoprotein receptor-/- mice.Arterioscler Thromb Vasc Biol. 2006 Aug;26(8):1814-20. doi: 10.1161/01.ATV.0000225289.30767.06. Epub 2006 May 4. Arterioscler Thromb Vasc Biol. 2006. PMID: 16675724
-
Dietary fat-induced alterations in atherosclerosis are abolished by ACAT2-deficiency in ApoB100 only, LDLr-/- mice.Arterioscler Thromb Vasc Biol. 2007 Jun;27(6):1396-402. doi: 10.1161/ATVBAHA.107.142802. Epub 2007 Apr 12. Arterioscler Thromb Vasc Biol. 2007. PMID: 17431188
-
Targeted depletion of hepatic ACAT2-driven cholesterol esterification reveals a non-biliary route for fecal neutral sterol loss.J Biol Chem. 2008 Apr 18;283(16):10522-34. doi: 10.1074/jbc.M707659200. Epub 2008 Feb 14. J Biol Chem. 2008. PMID: 18281279 Free PMC article.
-
Acyl coenzyme A: cholesterol acyltransferase inhibition and hepatic apolipoprotein B secretion.Clin Chim Acta. 1999 Aug;286(1-2):231-42. doi: 10.1016/s0009-8981(99)00104-7. Clin Chim Acta. 1999. PMID: 10511295 Review.
-
Acyl-coenzyme A:cholesteryl acyltransferase 2.Curr Opin Lipidol. 1999 Apr;10(2):89-95. doi: 10.1097/00041433-199904000-00002. Curr Opin Lipidol. 1999. PMID: 10327276 Review.
Cited by
-
The endoplasmic reticulum coat protein II transport machinery coordinates cellular lipid secretion and cholesterol biosynthesis.J Biol Chem. 2014 Feb 14;289(7):4244-61. doi: 10.1074/jbc.M113.479980. Epub 2013 Dec 13. J Biol Chem. 2014. PMID: 24338480 Free PMC article.
-
TG-interacting factor 1 acts as a transcriptional repressor of sterol O-acyltransferase 2.J Lipid Res. 2014 Apr;55(4):709-17. doi: 10.1194/jlr.M045922. Epub 2014 Jan 29. J Lipid Res. 2014. PMID: 24478032 Free PMC article.
-
Differential gene expression in bovine endometrial epithelial cells after challenge with LPS; specific implications for genes involved in embryo maternal interactions.PLoS One. 2019 Sep 5;14(9):e0222081. doi: 10.1371/journal.pone.0222081. eCollection 2019. PLoS One. 2019. PMID: 31487323 Free PMC article.
-
Acute sterol o-acyltransferase 2 (SOAT2) knockdown rapidly mobilizes hepatic cholesterol for fecal excretion.PLoS One. 2014 Jun 5;9(6):e98953. doi: 10.1371/journal.pone.0098953. eCollection 2014. PLoS One. 2014. PMID: 24901470 Free PMC article.
-
Multiple mechanisms limit the accumulation of unesterified cholesterol in the small intestine of mice deficient in both ACAT2 and ABCA1.Am J Physiol Gastrointest Liver Physiol. 2010 Nov;299(5):G1012-22. doi: 10.1152/ajpgi.00190.2010. Epub 2010 Aug 19. Am J Physiol Gastrointest Liver Physiol. 2010. PMID: 20724527 Free PMC article.
References
-
- Ludwig J., Viggiano T. R., McGill D. B., Oh B. J. (1980) Mayo Clin. Proc. 55, 434–438 - PubMed
-
- Varela-Rey M., Embade N., Ariz U., Lu S. C., Mato J. M., Martínez-Chantar M. L. (2009) Int. J. Biochem. Cell Biol. 41, 969–976 - PubMed
-
- Bradbury M. W. (2006) Am. J. Physiol. Gastrointest. Liver Physiol. 290, G194–G198 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
