

Epilepsy and the natural history of Rett syndrome
- PMID: 20231667
- PMCID: PMC2836870
- DOI: 10.1212/WNL.0b013e3181d6b852
Epilepsy and the natural history of Rett syndrome
Abstract
Background: Rett syndrome (RTT) is a neurodevelopmental disorder primarily seen in females, most with a mutation in MECP2. Epilepsy has been reported in 50%-80%. Previous reports were based on small sample sizes or parent-completed questionnaires, or failed to consider the impact of specific MECP2 mutations.
Methods: The Rare Disease Consortium Research Network for RTT is an NIH-funded project to characterize the clinical spectrum and natural history of RTT in advance of clinical trials. Evaluations include clinical status (classic vs atypical RTT), MECP2 mutations, clinical severity, and presence, frequency, and treatment of seizures.
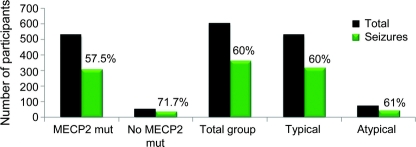
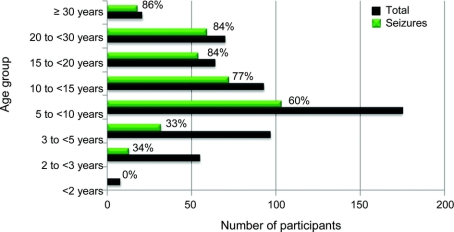
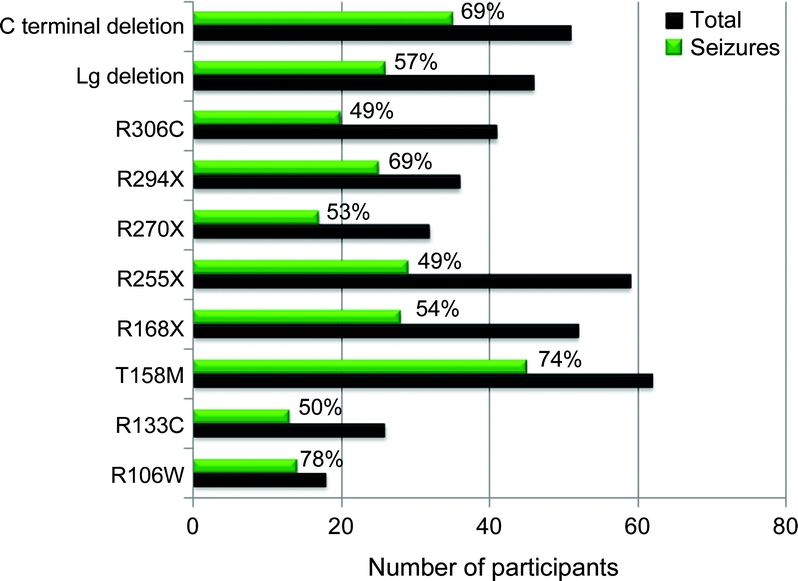
Results: Enrollment as of June 2008 is 602; 528 (88%) meet clinical criteria for classic RTT. Of these, 493 (93%) have MECP2 mutations. Age range was 8 months to 64 years. A total of 360 (60%) were reported to have seizures, including 315 (60%) classic and 45 (61%) atypical RTT. Physician assessment of the 602 indicated that 48% had seizures. There was no significant difference in seizure occurrence by race/ethnicity. A significant age impact for seizures was seen and seizures were infrequent before age 2 years. MECP2 mutations most frequently associated with epilepsy were T158M (74%) and R106W (78%), and less frequently R255X and R306C (both 49%). Individuals with seizures had greater overall clinical severity, and greater impairment of ambulation, hand use, and communication.
Discussion: Seizures are common in Rett syndrome, have an age-related onset and occurrence, vary by mutation, and are associated with greater clinical severity. This information represents a key consideration for designing clinical trials.
Figures
Similar articles
-
Epilepsy in Rett syndrome---the experience of a National Rett Center.Epilepsia. 2010 Jul;51(7):1252-8. doi: 10.1111/j.1528-1167.2010.02597.x. Epub 2010 May 13. Epilepsia. 2010. PMID: 20491871
-
Using a large international sample to investigate epilepsy in Rett syndrome.Dev Med Child Neurol. 2013 Jun;55(6):553-8. doi: 10.1111/dmcn.12093. Epub 2013 Feb 19. Dev Med Child Neurol. 2013. PMID: 23421866
-
Social impairments in Rett syndrome: characteristics and relationship with clinical severity.J Intellect Disabil Res. 2012 Mar;56(3):233-47. doi: 10.1111/j.1365-2788.2011.01404.x. Epub 2011 Mar 8. J Intellect Disabil Res. 2012. PMID: 21385260
-
Exploring the possible link between MeCP2 and oxidative stress in Rett syndrome.Free Radic Biol Med. 2015 Nov;88(Pt A):81-90. doi: 10.1016/j.freeradbiomed.2015.04.019. Epub 2015 May 8. Free Radic Biol Med. 2015. PMID: 25960047 Review.
-
The story of Rett syndrome: from clinic to neurobiology.Neuron. 2007 Nov 8;56(3):422-37. doi: 10.1016/j.neuron.2007.10.001. Neuron. 2007. PMID: 17988628 Review.
Cited by
-
Chloride imbalance in Fragile X syndrome.Front Neurosci. 2022 Oct 12;16:1008393. doi: 10.3389/fnins.2022.1008393. eCollection 2022. Front Neurosci. 2022. PMID: 36312023 Free PMC article.
-
Autism Spectrum Disorder with Epilepsy: A Research Protocol for a Clinical and Genetic Study.Genes (Basel). 2023 Dec 31;15(1):61. doi: 10.3390/genes15010061. Genes (Basel). 2023. PMID: 38254951 Free PMC article.
-
Consensus guidelines on managing Rett syndrome across the lifespan.BMJ Paediatr Open. 2020 Sep 13;4(1):e000717. doi: 10.1136/bmjpo-2020-000717. eCollection 2020. BMJ Paediatr Open. 2020. PMID: 32984552 Free PMC article.
-
Targeted pharmacological treatment of autism spectrum disorders: fragile X and Rett syndromes.Front Cell Neurosci. 2015 Feb 26;9:55. doi: 10.3389/fncel.2015.00055. eCollection 2015. Front Cell Neurosci. 2015. PMID: 25767435 Free PMC article. Review.
-
Growth failure and outcome in Rett syndrome: specific growth references.Neurology. 2012 Oct 16;79(16):1653-61. doi: 10.1212/WNL.0b013e31826e9a70. Epub 2012 Oct 3. Neurology. 2012. PMID: 23035069 Free PMC article.
References
-
- Hagberg B, Hanefield F, Percy A, Skjeldel O. An update on clinically applicable diagnostic criteria in Rett syndrome. Eur J Pediatr Neurol 2002;6:293–297. - PubMed
-
- Fitzgerald PM, Jankovic J, Glaze DG, Schultz RJ, Percy AK. Extrapyramidal involvement in Rett syndrome. Neurology 1990;40:293–295. - PubMed
-
- Nadia BB, Isabelle G, Rima N, et al. Parental view of epilepsy in Rett syndrome. Brain Dev 2008;30:126–130. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases