

Forced unbinding of GPR17 ligands from wild type and R255I mutant receptor models through a computational approach
- PMID: 20233425
- PMCID: PMC2850907
- DOI: 10.1186/1472-6807-10-8
Forced unbinding of GPR17 ligands from wild type and R255I mutant receptor models through a computational approach
Abstract
Background: GPR17 is a hybrid G-protein-coupled receptor (GPCR) activated by two unrelated ligand families, extracellular nucleotides and cysteinyl-leukotrienes (cysteinyl-LTs), and involved in brain damage and repair. Its exploitment as a target for novel neuro-reparative strategies depends on the elucidation of the molecular determinants driving binding of purinergic and leukotrienic ligands. Here, we applied docking and molecular dynamics simulations (MD) to analyse the binding and the forced unbinding of two GPR17 ligands (the endogenous purinergic agonist UDP and the leukotriene receptor antagonist pranlukast from both the wild-type (WT) receptor and a mutant model, where a basic residue hypothesized to be crucial for nucleotide binding had been mutated (R255I) to Ile.
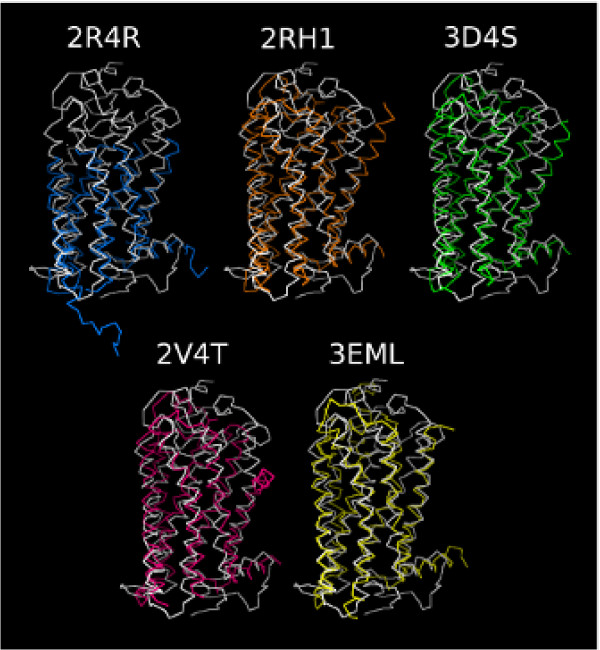
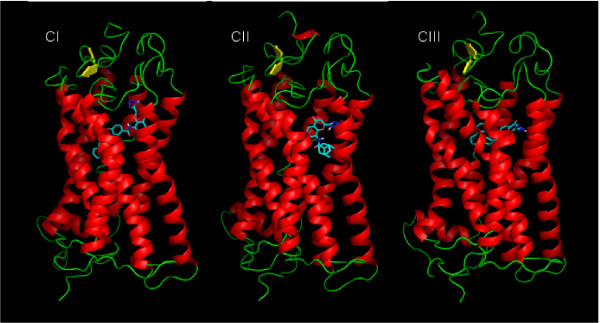
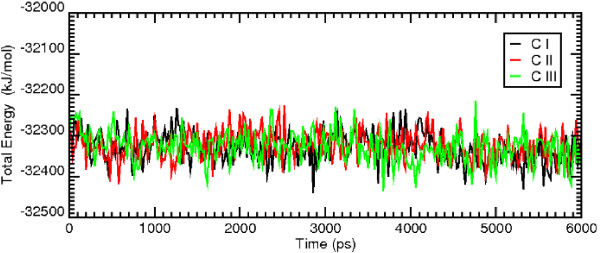
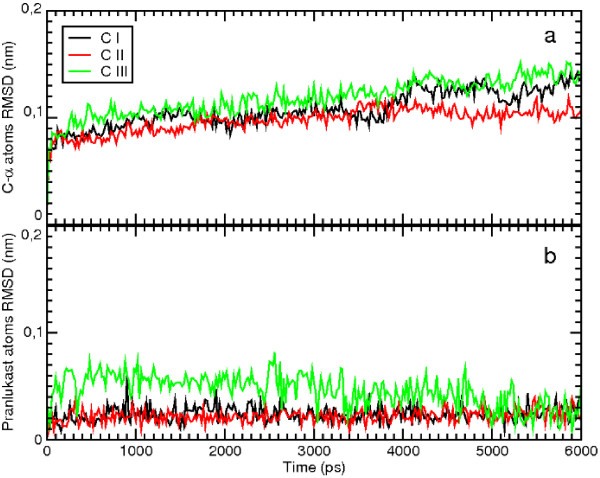
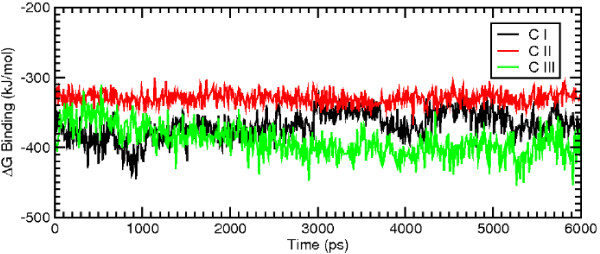
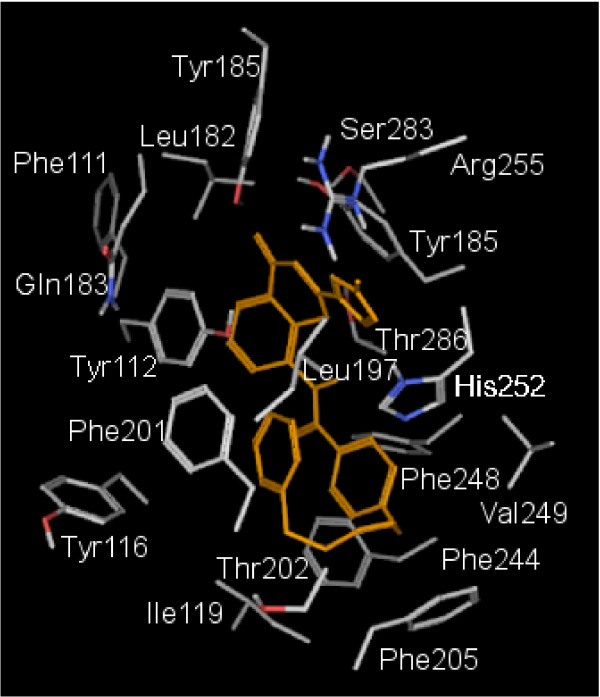
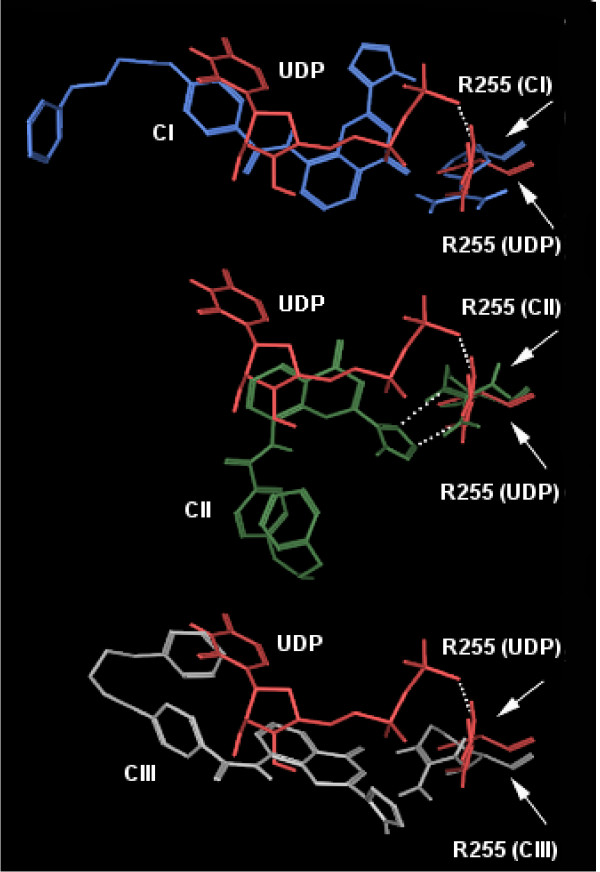
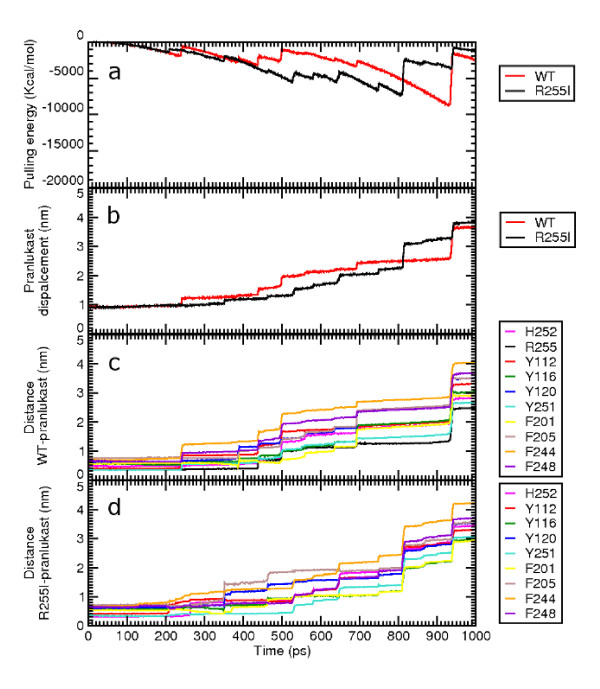
Results: MD suggested that GPR17 nucleotide binding pocket is enclosed between the helical bundle and extracellular loop (EL) 2. The driving interaction involves R255 and the UDP phosphate moiety. To support this hypothesis, steered MD experiments showed that the energy required to unbind UDP is higher for the WT receptor than for R255I. Three potential binding sites for pranlukast where instead found and analysed. In one of its preferential docking conformations, pranlukast tetrazole group is close to R255 and phenyl rings are placed into a subpocket highly conserved among GPCRs. Pulling forces developed to break polar and aromatic interactions of pranlukast were comparable. No differences between the WT receptor and the R255I receptor were found for the unbinding of pranlukast.
Conclusions: These data thus suggest that, in contrast to which has been hypothesized for nucleotides, the lack of the R255 residue doesn't affect the binding of pranlukast a crucial role for R255 in binding of nucleotides to GPR17. Aromatic interactions are instead likely to play a predominant role in the recognition of pranlukast, suggesting that two different binding subsites are present on GPR17.
Figures
Similar articles
-
GPR17: molecular modeling and dynamics studies of the 3-D structure and purinergic ligand binding features in comparison with P2Y receptors.BMC Bioinformatics. 2008 Jun 4;9:263. doi: 10.1186/1471-2105-9-263. BMC Bioinformatics. 2008. PMID: 18533035 Free PMC article.
-
Functional characterization of two isoforms of the P2Y-like receptor GPR17: [35S]GTPgammaS binding and electrophysiological studies in 1321N1 cells.Am J Physiol Cell Physiol. 2009 Oct;297(4):C1028-40. doi: 10.1152/ajpcell.00658.2008. Epub 2009 Jul 22. Am J Physiol Cell Physiol. 2009. PMID: 19625605
-
Agonist-induced desensitization/resensitization of human G protein-coupled receptor 17: a functional cross-talk between purinergic and cysteinyl-leukotriene ligands.J Pharmacol Exp Ther. 2011 Aug;338(2):559-67. doi: 10.1124/jpet.110.178715. Epub 2011 Apr 29. J Pharmacol Exp Ther. 2011. PMID: 21531793
-
The G Protein-Coupled Receptor GPR17: Overview and Update.ChemMedChem. 2016 Dec 6;11(23):2567-2574. doi: 10.1002/cmdc.201600453. Epub 2016 Nov 14. ChemMedChem. 2016. PMID: 27863043 Review.
-
Molecular mechanisms of ligand binding, signaling, and regulation within the superfamily of G-protein-coupled receptors: molecular modeling and mutagenesis approaches to receptor structure and function.Pharmacol Ther. 2004 Jul;103(1):21-80. doi: 10.1016/j.pharmthera.2004.05.002. Pharmacol Ther. 2004. PMID: 15251227 Review.
Cited by
-
Exploring the free-energy landscapes of biological systems with steered molecular dynamics.Phys Chem Chem Phys. 2011 Apr 7;13(13):6176-83. doi: 10.1039/c0cp02799e. Epub 2011 Feb 25. Phys Chem Chem Phys. 2011. PMID: 21359274 Free PMC article.
-
The regulated expression, intracellular trafficking, and membrane recycling of the P2Y-like receptor GPR17 in Oli-neu oligodendroglial cells.J Biol Chem. 2013 Feb 15;288(7):5241-56. doi: 10.1074/jbc.M112.404996. Epub 2013 Jan 3. J Biol Chem. 2013. PMID: 23288840 Free PMC article.
-
Intestinal Gpr17 deficiency improves glucose metabolism by promoting GLP-1 secretion.Cell Rep. 2022 Jan 4;38(1):110179. doi: 10.1016/j.celrep.2021.110179. Cell Rep. 2022. PMID: 34986353 Free PMC article.
-
Gastrin-releasing peptide/neuromedin B receptor antagonists PD176252, PD168368, and related analogs are potent agonists of human formyl-peptide receptors.Mol Pharmacol. 2011 Jan;79(1):77-90. doi: 10.1124/mol.110.068288. Epub 2010 Oct 13. Mol Pharmacol. 2011. PMID: 20943772 Free PMC article.
-
Abnormal Upregulation of GPR17 Receptor Contributes to Oligodendrocyte Dysfunction in SOD1 G93A Mice.Int J Mol Sci. 2020 Mar 31;21(7):2395. doi: 10.3390/ijms21072395. Int J Mol Sci. 2020. PMID: 32244295 Free PMC article.
References
-
- Abbracchio MP, Burnstock G, Boeynaems J, Barnard EA, Boyer J, Kennedy C, Knight GE, Fumagalli M, Gachet C, Jacobson KA, Weisman GA. International Union of Pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacol Rev. 2006;58:281–341. doi: 10.1124/pr.58.3.3. - DOI - PMC - PubMed
