

Transcriptome sequencing in an ecologically important tree species: assembly, annotation, and marker discovery
- PMID: 20233449
- PMCID: PMC2851599
- DOI: 10.1186/1471-2164-11-180
Transcriptome sequencing in an ecologically important tree species: assembly, annotation, and marker discovery
Abstract
Background: Massively parallel sequencing of cDNA is now an efficient route for generating enormous sequence collections that represent expressed genes. This approach provides a valuable starting point for characterizing functional genetic variation in non-model organisms, especially where whole genome sequencing efforts are currently cost and time prohibitive. The large and complex genomes of pines (Pinus spp.) have hindered the development of genomic resources, despite the ecological and economical importance of the group. While most genomic studies have focused on a single species (P. taeda), genomic level resources for other pines are insufficiently developed to facilitate ecological genomic research. Lodgepole pine (P. contorta) is an ecologically important foundation species of montane forest ecosystems and exhibits substantial adaptive variation across its range in western North America. Here we describe a sequencing study of expressed genes from P. contorta, including their assembly and annotation, and their potential for molecular marker development to support population and association genetic studies.
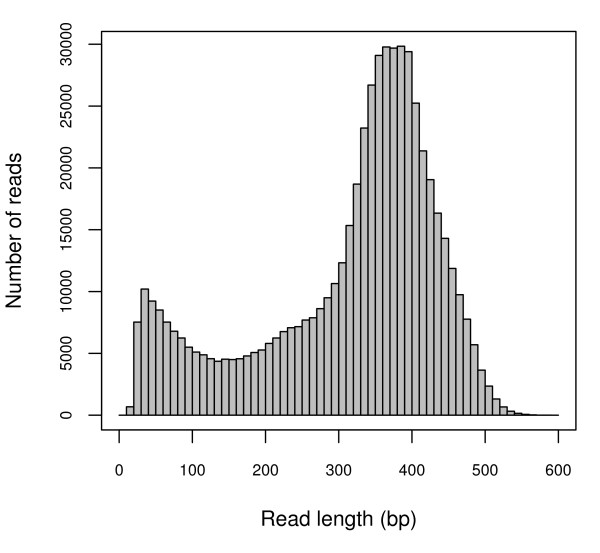
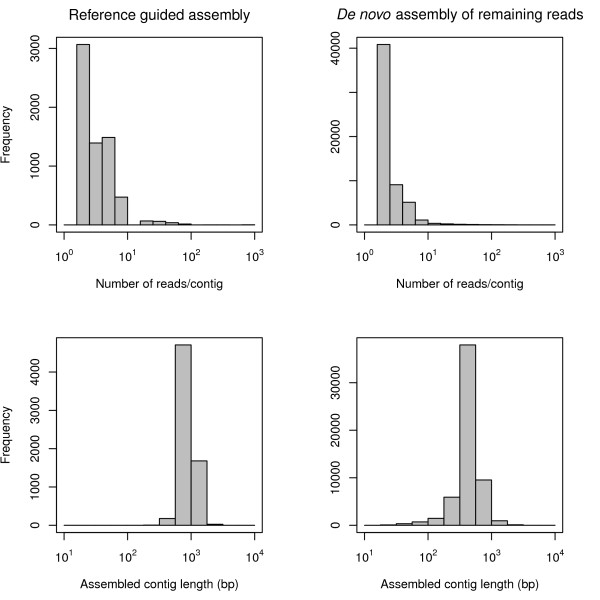
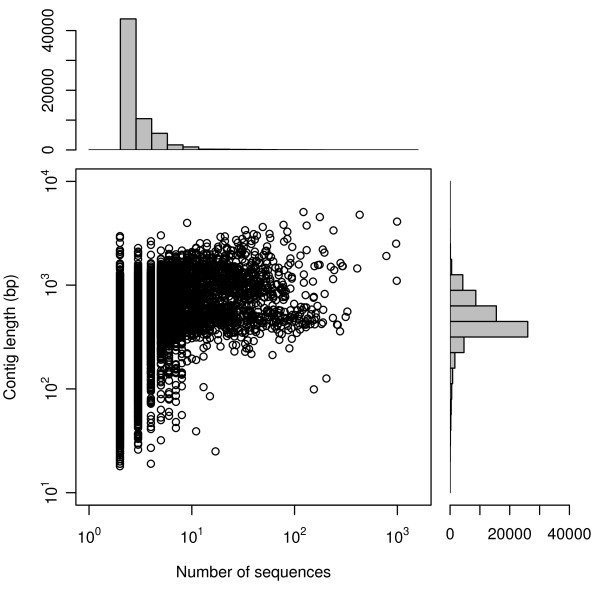
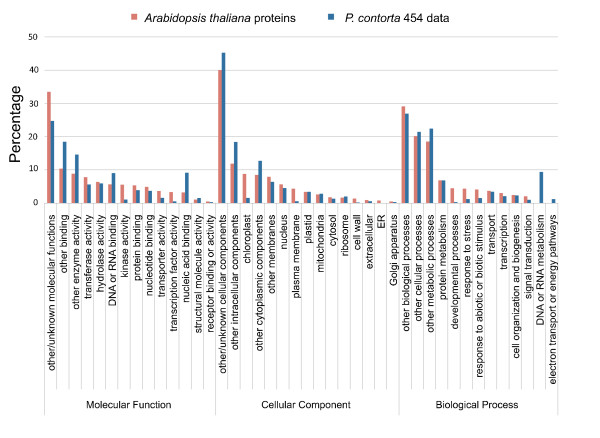
Results: We obtained 586,732 sequencing reads from a 454 GS XLR70 Titanium pyrosequencer (mean length: 306 base pairs). A combination of reference-based and de novo assemblies yielded 63,657 contigs, with 239,793 reads remaining as singletons. Based on sequence similarity with known proteins, these sequences represent approximately 17,000 unique genes, many of which are well covered by contig sequences. This sequence collection also included a surprisingly large number of retrotransposon sequences, suggesting that they are highly transcriptionally active in the tissues we sampled. We located and characterized thousands of simple sequence repeats and single nucleotide polymorphisms as potential molecular markers in our assembled and annotated sequences. High quality PCR primers were designed for a substantial number of the SSR loci, and a large number of these were amplified successfully in initial screening.
Conclusions: This sequence collection represents a major genomic resource for P. contorta, and the large number of genetic markers characterized should contribute to future research in this and other pines. Our results illustrate the utility of next generation sequencing as a basis for marker development and population genomics in non-model species.
Figures

Similar articles
-
Combined de novo and genome guided assembly and annotation of the Pinus patula juvenile shoot transcriptome.BMC Genomics. 2015 Dec 12;16:1057. doi: 10.1186/s12864-015-2277-7. BMC Genomics. 2015. PMID: 26652261 Free PMC article.
-
Generation, functional annotation and comparative analysis of black spruce (Picea mariana) ESTs: an important conifer genomic resource.BMC Genomics. 2013 Oct 11;14:702. doi: 10.1186/1471-2164-14-702. BMC Genomics. 2013. PMID: 24119028 Free PMC article.
-
De novo assembly of maritime pine transcriptome: implications for forest breeding and biotechnology.Plant Biotechnol J. 2014 Apr;12(3):286-99. doi: 10.1111/pbi.12136. Epub 2013 Nov 21. Plant Biotechnol J. 2014. PMID: 24256179
-
Application of large-scale sequencing to marker discovery in plants.J Biosci. 2012 Nov;37(5):829-41. doi: 10.1007/s12038-012-9253-z. J Biosci. 2012. PMID: 23107919 Review.
-
Forest-tree population genomics and adaptive evolution.New Phytol. 2006;170(2):227-38. doi: 10.1111/j.1469-8137.2006.01686.x. New Phytol. 2006. PMID: 16608450 Review.
Cited by
-
SNP markers and their impact on plant breeding.Int J Plant Genomics. 2012;2012:728398. doi: 10.1155/2012/728398. Epub 2012 Dec 18. Int J Plant Genomics. 2012. PMID: 23316221 Free PMC article.
-
High-throughput sequencing discovery of conserved and novel microRNAs in Chinese cabbage (Brassica rapa L. ssp. pekinensis).Mol Genet Genomics. 2012 Jul;287(7):555-63. doi: 10.1007/s00438-012-0699-3. Epub 2012 May 29. Mol Genet Genomics. 2012. PMID: 22643909
-
Evolutionary insights from comparative transcriptome and transcriptome-wide coalescence analyses in Tetrastigma hemsleyanum.BMC Plant Biol. 2018 Sep 24;18(1):208. doi: 10.1186/s12870-018-1429-8. BMC Plant Biol. 2018. PMID: 30249188 Free PMC article.
-
Functional genomics by integrated analysis of transcriptome of sweet potato (Ipomoea batatas (L.) Lam.) during root formation.Genes Genomics. 2020 May;42(5):581-596. doi: 10.1007/s13258-020-00927-7. Epub 2020 Apr 2. Genes Genomics. 2020. PMID: 32240514
-
De novo assembly of transcriptomes, mining, and development of novel EST-SSR markers in Curcuma alismatifolia (Zingiberaceae family) through Illumina sequencing.Sci Rep. 2019 Feb 28;9(1):3047. doi: 10.1038/s41598-019-39944-2. Sci Rep. 2019. PMID: 30816255 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
