

Chemotherapy enhances tumor cell susceptibility to CTL-mediated killing during cancer immunotherapy in mice
- PMID: 20234093
- PMCID: PMC2846048
- DOI: 10.1172/JCI40269
Chemotherapy enhances tumor cell susceptibility to CTL-mediated killing during cancer immunotherapy in mice
Abstract
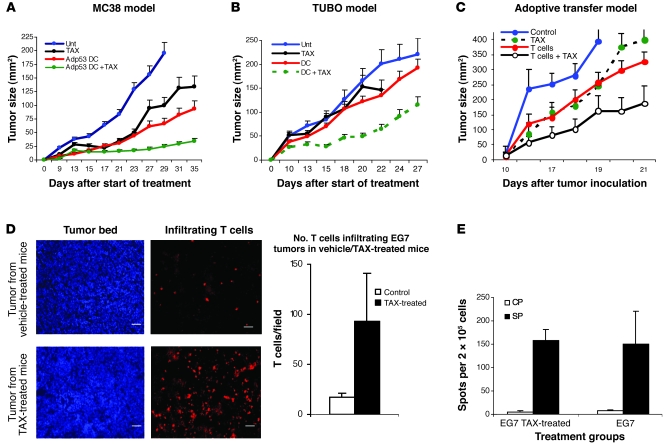
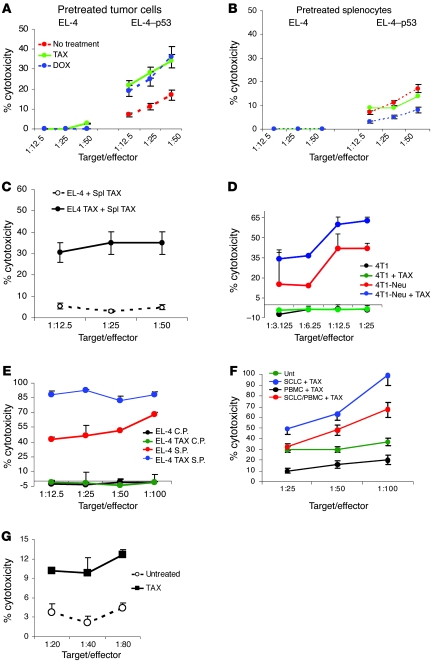
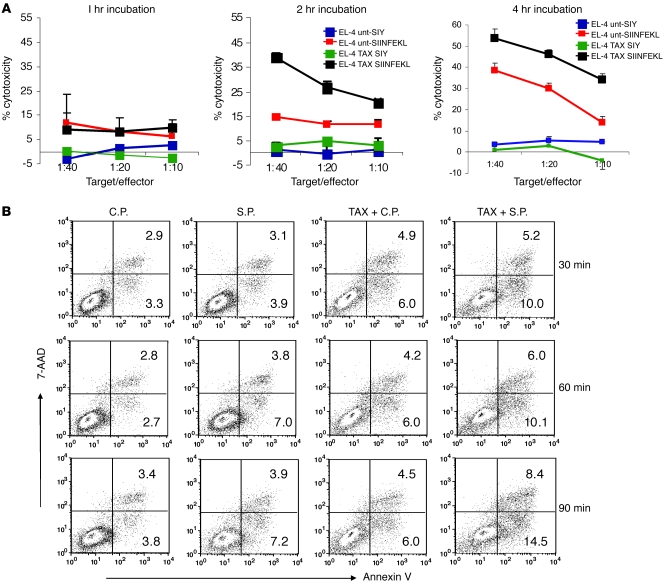
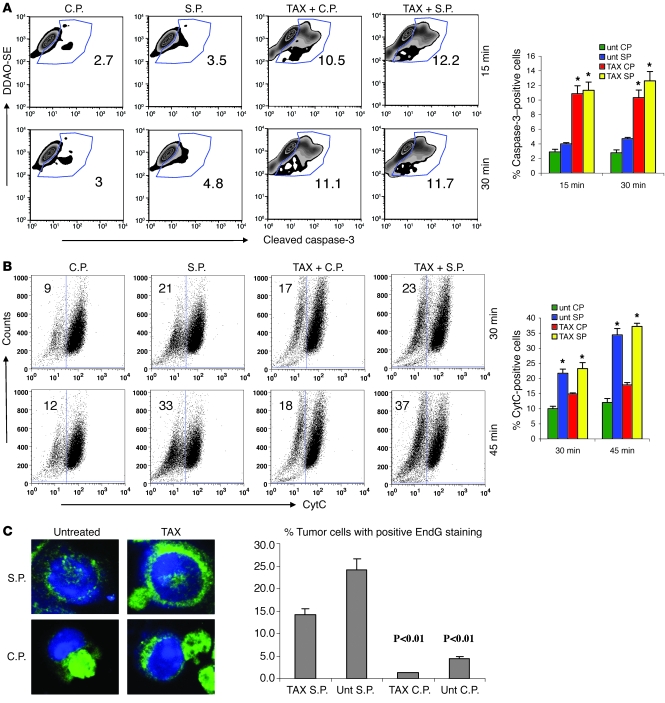
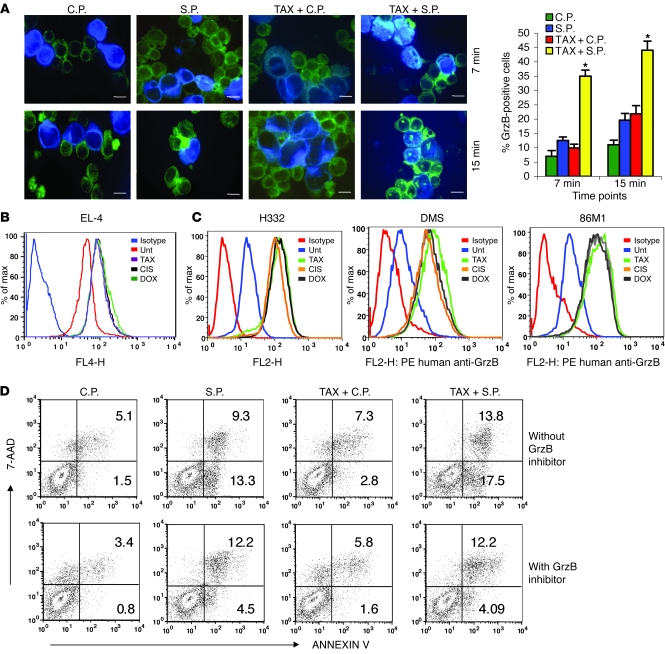
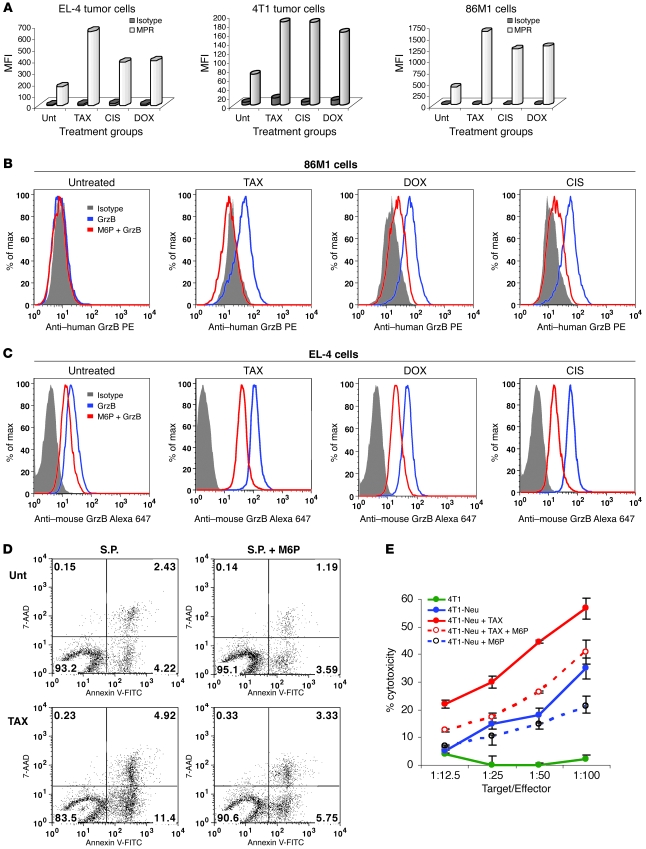
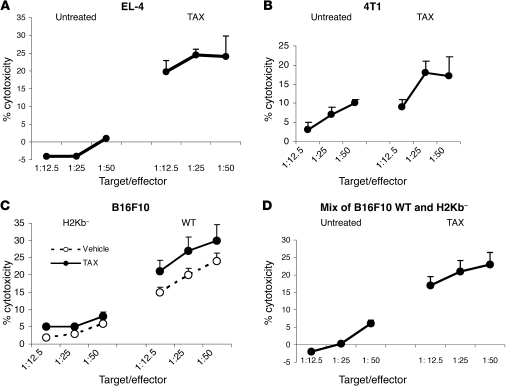
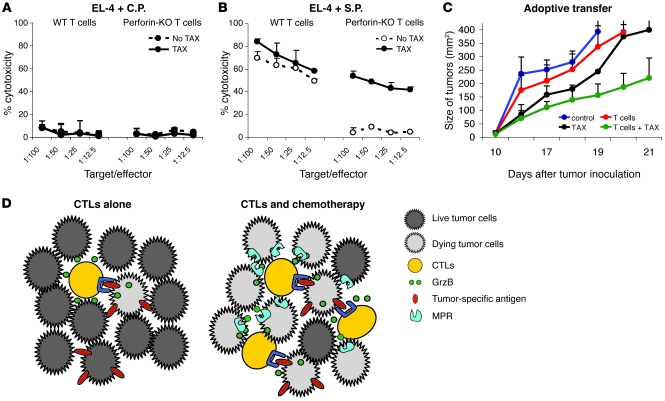
Cancer immunotherapy faces a serious challenge because of low clinical efficacy. Recently, a number of clinical studies have reported the serendipitous finding of high rates of objective clinical response when cancer vaccines are combined with chemotherapy in patients with different types of cancers. However, the mechanism of this phenomenon remains unclear. Here, we tested in mice several cancer vaccines and an adoptive T cell transfer approach to cancer immunotherapy in combination with several widely used chemotherapeutic drugs. We found that chemotherapy made tumor cells more susceptible to the cytotoxic effect of CTLs through a dramatic perforin-independent increase in permeability to GrzB released by the CTLs. This effect was mediated via upregulation of mannose-6-phosphate receptors on the surface of tumor cells and was observed in mouse and human cells. When combined with chemotherapy, CTLs raised against specific antigens were able to induce apoptosis in neighboring tumor cells that did not express those antigens. These data suggest that small numbers of CTLs could mediate a potent antitumor effect when combined with chemotherapy. In addition, these results provide a strong rationale for combining these modalities for the treatment of patients with advanced cancers.
Figures
Comment in
-
Chemotherapy. Conventional chemotherapy boosts the effect of cancer vaccines.Nat Rev Clin Oncol. 2010 Jun;7(6):297. doi: 10.1038/nrclinonc.2010.73. Nat Rev Clin Oncol. 2010. PMID: 20527681 No abstract available.
Similar articles
-
Chemotherapeutic drugs may be used to enhance the killing efficacy of human tumor antigen peptide-specific CTLs.J Immunother. 2008 Feb-Mar;31(2):132-47. doi: 10.1097/CJI.0b013e31815b69c8. J Immunother. 2008. PMID: 18481383
-
Chemosensitization of human prostate carcinoma cell lines to anti-fas-mediated cytotoxicity and apoptosis.Clin Cancer Res. 1997 Jun;3(6):963-72. Clin Cancer Res. 1997. PMID: 9815772
-
Rapid deletion and inactivation of CTLs upon recognition of a number of target cells over a critical threshold.J Immunol. 2013 Oct 1;191(7):3534-44. doi: 10.4049/jimmunol.1300803. Epub 2013 Sep 9. J Immunol. 2013. PMID: 24018271
-
Multiphoton imaging of cytotoxic T lymphocyte-mediated antitumor immune responses.Curr Top Microbiol Immunol. 2009;334:265-87. doi: 10.1007/978-3-540-93864-4_11. Curr Top Microbiol Immunol. 2009. PMID: 19521689 Review.
-
Adoptive immunotherapy in combination with chemotherapy for cancer treatment.Prog Exp Tumor Res. 1988;32:128-53. doi: 10.1159/000414676. Prog Exp Tumor Res. 1988. PMID: 3287448 Review. No abstract available.
Cited by
-
Novel mechanism of synergistic effects of conventional chemotherapy and immune therapy of cancer.Cancer Immunol Immunother. 2013 Mar;62(3):405-10. doi: 10.1007/s00262-012-1390-6. Epub 2013 Feb 20. Cancer Immunol Immunother. 2013. PMID: 23423351 Free PMC article. Review.
-
Engineered drug resistant γδ T cells kill glioblastoma cell lines during a chemotherapy challenge: a strategy for combining chemo- and immunotherapy.PLoS One. 2013;8(1):e51805. doi: 10.1371/journal.pone.0051805. Epub 2013 Jan 11. PLoS One. 2013. PMID: 23326319 Free PMC article.
-
Electroporation driven delivery of both an IL-12 expressing plasmid and cisplatin synergizes to inhibit B16 melanoma tumor growth through an NK cell mediated tumor killing mechanism.Hum Vaccin Immunother. 2012 Nov 1;8(11):1714-21. doi: 10.4161/hv.22346. Epub 2012 Nov 1. Hum Vaccin Immunother. 2012. PMID: 23151450 Free PMC article.
-
Neoadjuvant Chemotherapy Increases Cytotoxic T Cell, Tissue Resident Memory T Cell, and B Cell Infiltration in Resectable NSCLC.J Thorac Oncol. 2021 Jan;16(1):127-139. doi: 10.1016/j.jtho.2020.09.027. Epub 2020 Oct 21. J Thorac Oncol. 2021. PMID: 33096269 Free PMC article.
-
Implication of tumor microenvironment in chemoresistance: tumor-associated stromal cells protect tumor cells from cell death.Int J Mol Sci. 2012;13(8):9545-9571. doi: 10.3390/ijms13089545. Epub 2012 Jul 30. Int J Mol Sci. 2012. PMID: 22949815 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
