

Practically useful: what the Rosetta protein modeling suite can do for you
- PMID: 20235548
- PMCID: PMC2850155
- DOI: 10.1021/bi902153g
Practically useful: what the Rosetta protein modeling suite can do for you
Abstract
The objective of this review is to enable researchers to use the software package Rosetta for biochemical and biomedicinal studies. We provide a brief review of the six most frequent research problems tackled with Rosetta. For each of these six tasks, we provide a tutorial that illustrates a basic Rosetta protocol. The Rosetta method was originally developed for de novo protein structure prediction and is regularly one of the best performers in the community-wide biennial Critical Assessment of Structure Prediction. Predictions for protein domains with fewer than 125 amino acids regularly have a backbone root-mean-square deviation of better than 5.0 A. More impressively, there are several cases in which Rosetta has been used to predict structures with atomic level accuracy better than 2.5 A. In addition to de novo structure prediction, Rosetta also has methods for molecular docking, homology modeling, determining protein structures from sparse experimental NMR or EPR data, and protein design. Rosetta has been used to accurately design a novel protein structure, predict the structure of protein-protein complexes, design altered specificity protein-protein and protein-DNA interactions, and stabilize proteins and protein complexes. Most recently, Rosetta has been used to solve the X-ray crystallographic phase problem.
Figures
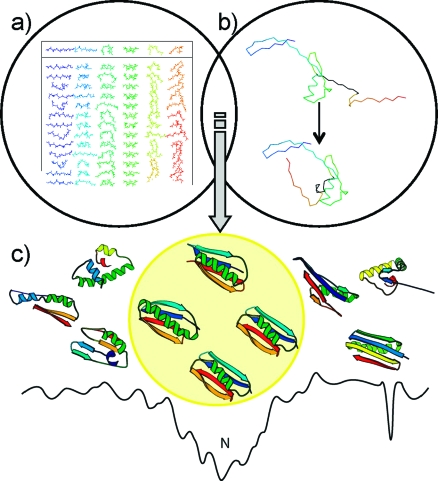
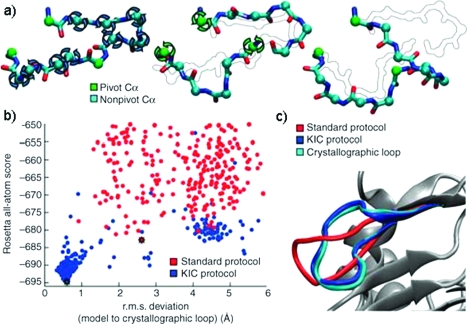
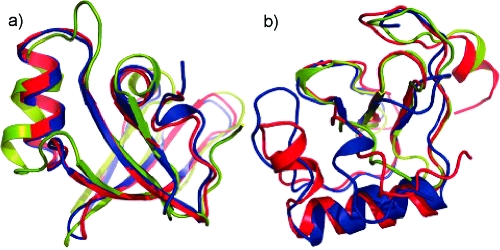


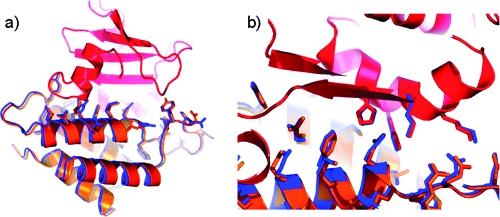


References
-
- Berman H. M.; Battistuz T.; Bhat T. N.; Bluhm W. F.; Bourne P. E.; Burkhardt K.; Feng Z.; Gilliland G. L.; Iype L.; Jain S.; Fagan P.; Marvin J.; Padilla D.; Ravichandran V.; Schneider B.; Thanki N.; Weissig H.; Westbrook J. D.; Zardecki C. (2002) The Protein Data Bank. Acta Crystallogr. D58, 899–907. - PubMed
-
- Bernstein F. C.; Koetzle T. F.; Williams G. J.; Meyer E. F. Jr.; Brice M. D.; Rodgers J. R.; Kennard O.; Shimanouchi T.; Tasumi M. (1977) The Protein Data Bank: A computer-based archival file for macromolecular structures. J. Mol. Biol. 112, 535–542. - PubMed
-
- Wang C.; Bradley P.; Baker D. (2007) Protein-protein docking with backbone flexibility. J. Mol. Biol. 373, 503–519. - PubMed
-
- Simons K. T.; Kooperberg C.; Huang E.; Baker D. (1997) Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and Bayesian scoring functions. J. Mol. Biol. 268, 209–225. - PubMed
-
- Bystroff C.; Simons K. T.; Han K. F.; Baker D. (1996) Local sequence-structure correlations in proteins. Curr. Opin. Biotechnol. 7, 417–421. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
