

VEGF-A165b is cytoprotective and antiangiogenic in the retina
- PMID: 20237249
- PMCID: PMC2910648
- DOI: 10.1167/iovs.09-4296
VEGF-A165b is cytoprotective and antiangiogenic in the retina
Abstract
Purpose: A number of key ocular diseases, including diabetic retinopathy and age-related macular degeneration, are characterized by localized areas of epithelial or endothelial damage, which can ultimately result in the growth of fragile new blood vessels, vitreous hemorrhage, and retinal detachment. VEGF-A(165), the principal neovascular agent in ocular angiogenic conditions, is formed by proximal splice site selection in its terminal exon 8. Alternative splicing of this exon results in an antiangiogenic isoform, VEGF-A(165)b, which is downregulated in diabetic retinopathy. Here the authors investigate the antiangiogenic activity of VEGF(165)b and its effect on retinal epithelial and endothelial cell survival.
Methods: VEGF-A(165)b was injected intraocularly in a mouse model of retinal neovascularization (oxygen-induced retinopathy [OIR]). Cytotoxicity and cell migration assays were used to determine the effect of VEGF-A(165)b.
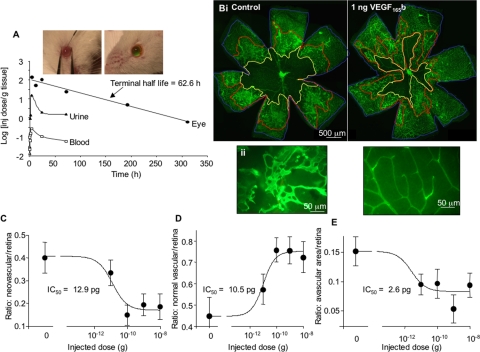
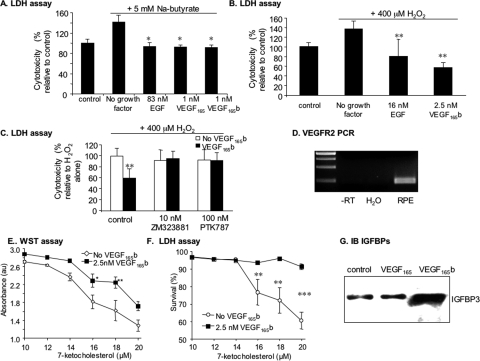
Results: VEGF-A(165)b dose dependently inhibited angiogenesis (IC(50), 12.6 pg/eye) and retinal endothelial migration induced by 1 nM VEGF-A(165) across monolayers in culture (IC(50), 1 nM). However, it also acts as a survival factor for endothelial cells and retinal epithelial cells through VEGFR2 and can stimulate downstream signaling. Furthermore, VEGF-A(165)b injection, while inhibiting neovascular proliferation in the eye, reduced the ischemic insult in OIR (IC(50), 2.6 pg/eye). Unlike bevacizumab, pegaptanib did not interact directly with VEGF-A(165)b.
Conclusions: The survival effects of VEGF-A(165)b signaling can protect the retina from ischemic damage. These results suggest that VEGF-A(165)b may be a useful therapeutic agent in ischemia-induced angiogenesis and a cytoprotective agent for retinal pigment epithelial cells.
Figures




Similar articles
-
Recombinant human VEGF165b inhibits experimental choroidal neovascularization.Invest Ophthalmol Vis Sci. 2010 Aug;51(8):4282-8. doi: 10.1167/iovs.09-4360. Epub 2010 Mar 17. Invest Ophthalmol Vis Sci. 2010. PMID: 20237252 Free PMC article.
-
Antiangiogenic effect of betaine on pathologic retinal neovascularization via suppression of reactive oxygen species mediated vascular endothelial growth factor signaling.Vascul Pharmacol. 2017 Mar;90:19-26. doi: 10.1016/j.vph.2016.07.007. Epub 2016 Jul 27. Vascul Pharmacol. 2017. PMID: 27473515
-
Vascular endothelial growth factor-A165b ameliorates outer-retinal barrier and vascular dysfunction in the diabetic retina.Clin Sci (Lond). 2017 Jun 1;131(12):1225-1243. doi: 10.1042/CS20170102. Epub 2017 Mar 24. Clin Sci (Lond). 2017. PMID: 28341661 Free PMC article.
-
Anti-vascular endothelial growth factor for neovascular age-related macular degeneration.Cochrane Database Syst Rev. 2019 Mar 4;3(3):CD005139. doi: 10.1002/14651858.CD005139.pub4. Cochrane Database Syst Rev. 2019. PMID: 30834517 Free PMC article.
-
Molecular diversity of VEGF-A as a regulator of its biological activity.Microcirculation. 2009 Oct;16(7):572-92. doi: 10.1080/10739680902997333. Epub 2009 Jun 1. Microcirculation. 2009. PMID: 19521900 Free PMC article. Review.
Cited by
-
Antiangiogenic VEGF isoform in inflammatory myopathies.Mediators Inflamm. 2013;2013:219313. doi: 10.1155/2013/219313. Epub 2013 Jun 12. Mediators Inflamm. 2013. PMID: 23840094 Free PMC article.
-
The VEGF-A exon 8 splicing-sensitive fluorescent reporter mouse is a novel tool to assess the effects of splicing regulatory compounds in vivo.RNA Biol. 2019 Dec;16(12):1672-1681. doi: 10.1080/15476286.2019.1652522. Epub 2019 Aug 21. RNA Biol. 2019. PMID: 31432737 Free PMC article.
-
Recombinant human VEGF165b inhibits experimental choroidal neovascularization.Invest Ophthalmol Vis Sci. 2010 Aug;51(8):4282-8. doi: 10.1167/iovs.09-4360. Epub 2010 Mar 17. Invest Ophthalmol Vis Sci. 2010. PMID: 20237252 Free PMC article.
-
VEGF spliced variants: possible role of anti-angiogenesis therapy.J Nucleic Acids. 2012;2012:162692. doi: 10.1155/2012/162692. Epub 2011 Oct 13. J Nucleic Acids. 2012. PMID: 22013509 Free PMC article.
-
Vascular endothelial growth factor-165b protects the blood-retinal barrier from damage after acute high intraocular pressure in rats.Int J Ophthalmol. 2022 Aug 18;15(8):1231-1239. doi: 10.18240/ijo.2022.08.02. eCollection 2022. Int J Ophthalmol. 2022. PMID: 36017048 Free PMC article.
References
-
- Roth F, Bindewald A, Holz FG. Key pathophysiologic pathways in age-related macular disease. Graefes Arch Clin Exp Ophthalmol 2004;242:710–716 - PubMed
-
- Kinose F, Roscilli G, Lamartina S, et al. Inhibition of retinal and choroidal neovascularization by a novel KDR kinase inhibitor. Mol Vis 2005;11:366–373 - PubMed
-
- Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 1992;359:843–845 - PubMed
-
- Rosenfeld PJ, Brown DM, Heier JS, et al. Ranibizumab for neovascular age-related macular degeneration. N Engl J Med 2006;355:1419–1431 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
