

MISHIMA--a new method for high speed multiple alignment of nucleotide sequences of bacterial genome scale data
- PMID: 20298584
- PMCID: PMC2848238
- DOI: 10.1186/1471-2105-11-142
MISHIMA--a new method for high speed multiple alignment of nucleotide sequences of bacterial genome scale data
Abstract
Background: Large nucleotide sequence datasets are becoming increasingly common objects of comparison. Complete bacterial genomes are reported almost everyday. This creates challenges for developing new multiple sequence alignment methods. Conventional multiple alignment methods are based on pairwise alignment and/or progressive alignment techniques. These approaches have performance problems when the number of sequences is large and when dealing with genome scale sequences.
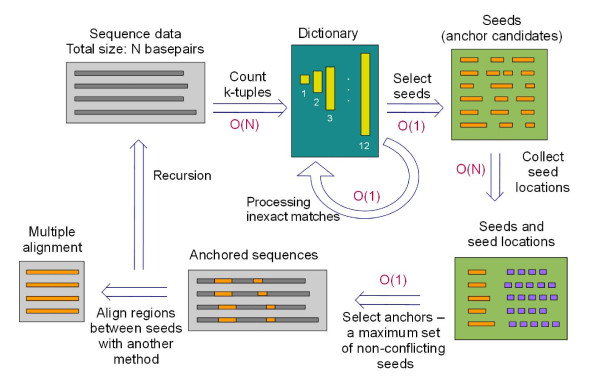
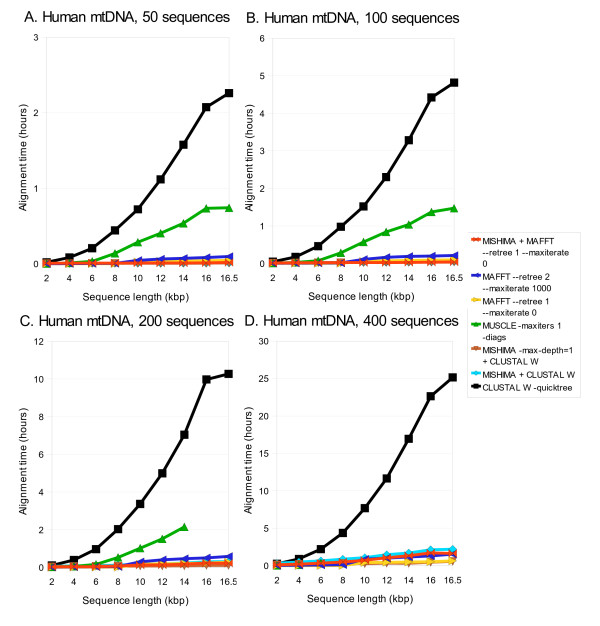
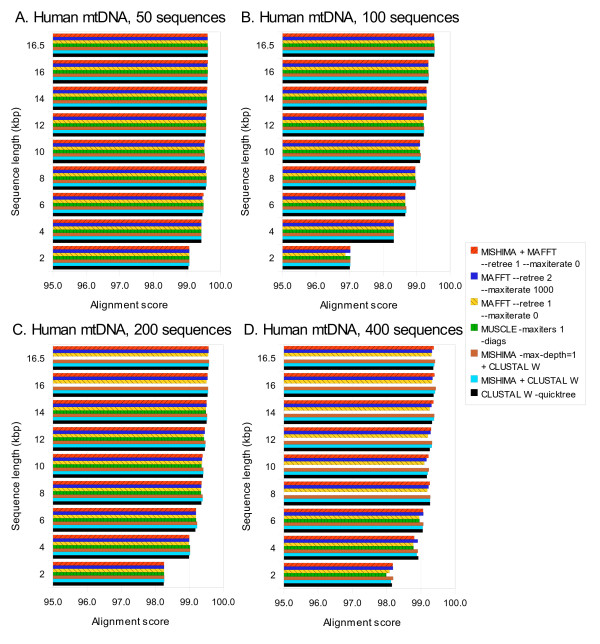
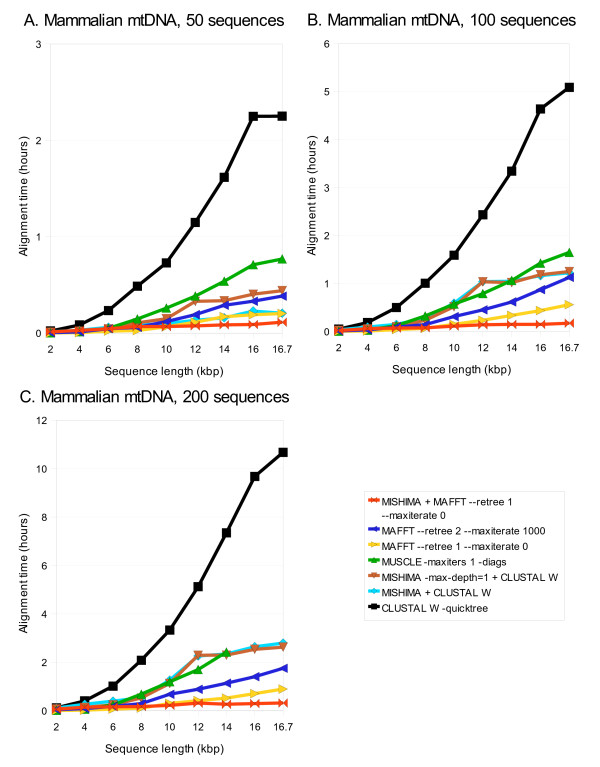
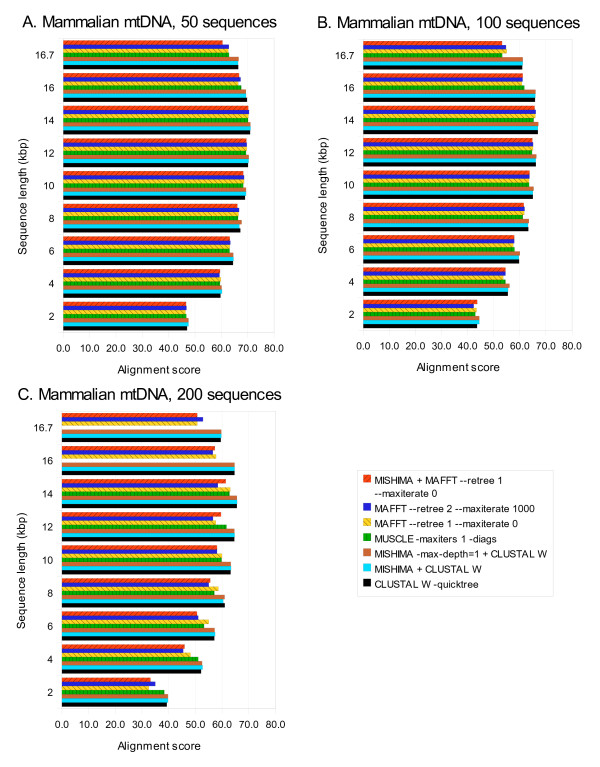
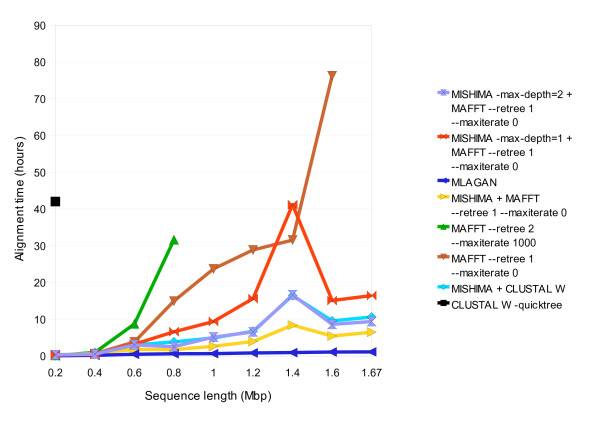
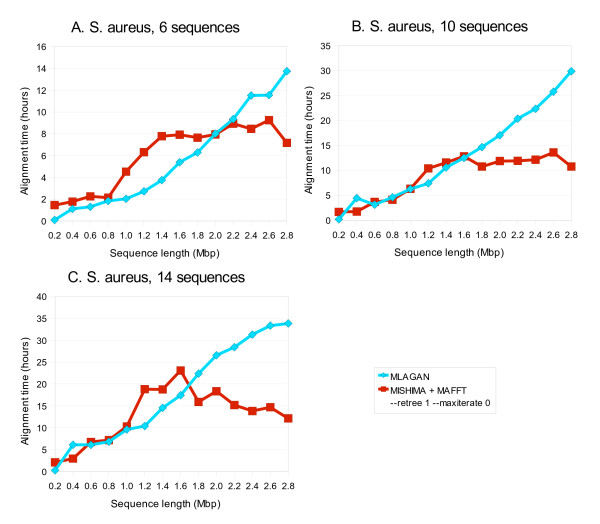
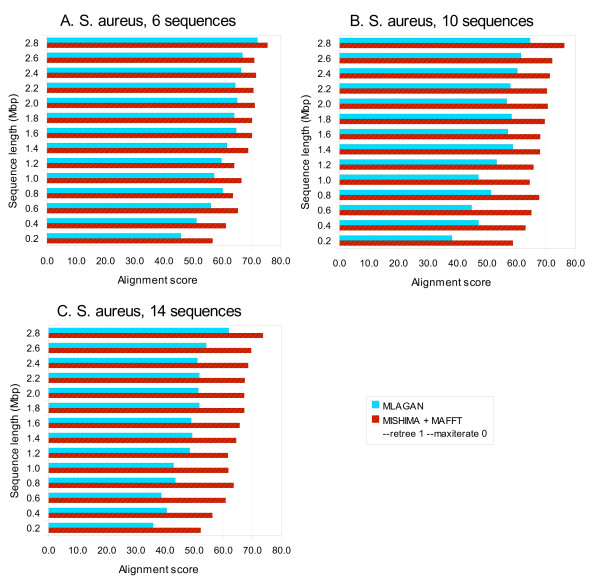
Results: We present a new method of multiple sequence alignment, called MISHIMA (Method for Inferring Sequence History In terms of Multiple Alignment), that does not depend on pairwise sequence comparison. A new algorithm is used to quickly find rare oligonucleotide sequences shared by all sequences. Divide and conquer approach is then applied to break the sequences into fragments that can be aligned independently by an external alignment program. These partial alignments are assembled together to form a complete alignment of the original sequences.
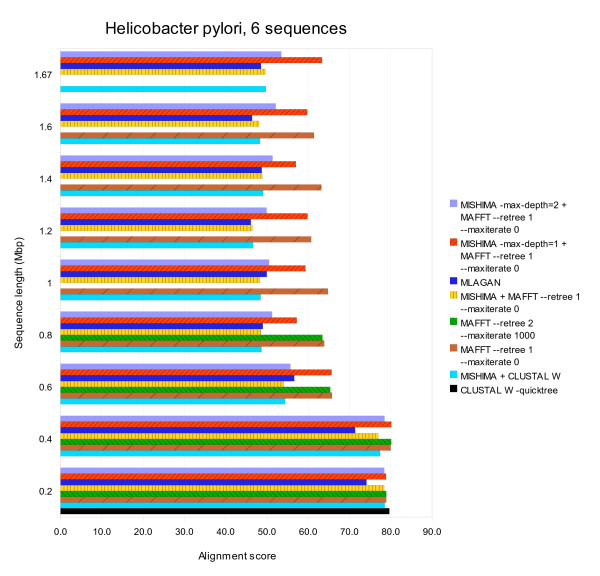
Conclusions: MISHIMA provides improved performance compared to the commonly used multiple alignment methods. As an example, six complete genome sequences of bacteria species Helicobacter pylori (about 1.7 Mb each) were successfully aligned in about 6 hours using a single PC.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
