

High-resolution detection of identity by descent in unrelated individuals
- PMID: 20303063
- PMCID: PMC2850444
- DOI: 10.1016/j.ajhg.2010.02.021
High-resolution detection of identity by descent in unrelated individuals
Abstract
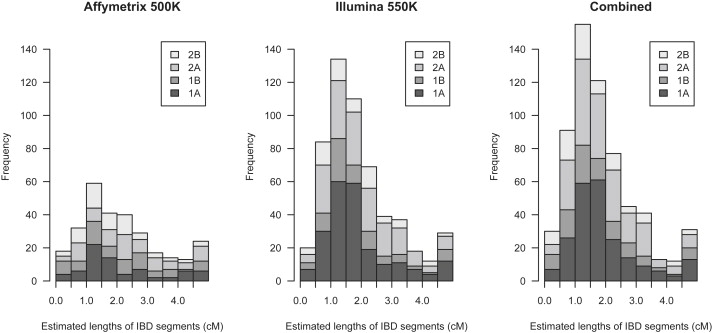
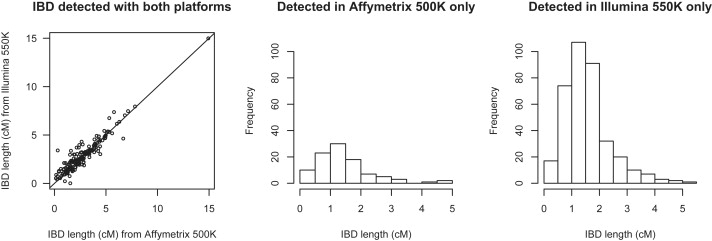
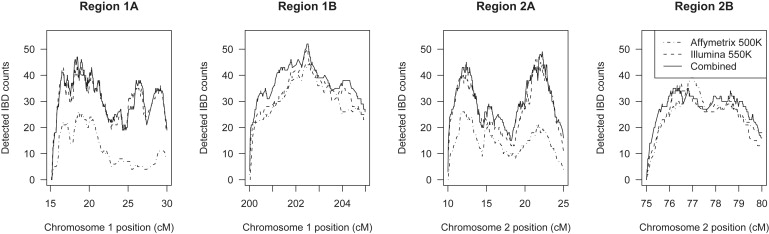
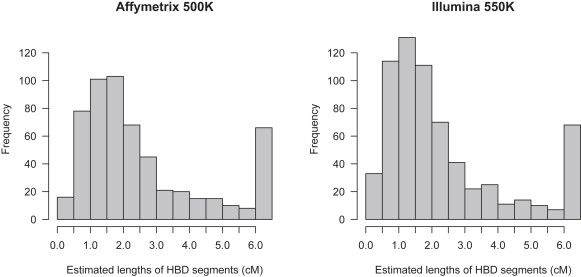
Detection of recent identity by descent (IBD) in population samples is important for population-based linkage mapping and for highly accurate genotype imputation and haplotype-phase inference. We present a method for detection of recent IBD in population samples. Our method accounts for linkage disequilibrium between SNPs to enable full use of high-density SNP data. We find that our method can detect segments of a length of 2 cM with moderate power and negligible false discovery rate in Illumina 550K data in Northwestern Europeans. We compare our method with GERMLINE and PLINK, and we show that our method has a level of resolution that is significantly better than these existing methods, thus extending the usefulness of recent IBD in analysis of high-density SNP data. We survey four genomic regions in a sample of UK individuals of European descent and find that on average, at a given location, our method detects IBD in 2.7 per 10,000 pairs of individuals in Illumina 550K data. We also present methodology and results for detection of homozygosity by descent (HBD) and survey the whole genome in a sample of 1373 UK individuals of European descent. We detect HBD in 4.7 individuals per 10,000 on average at a given location. Our methodology is implemented in the freely available BEAGLE software package.
(c) 2010 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
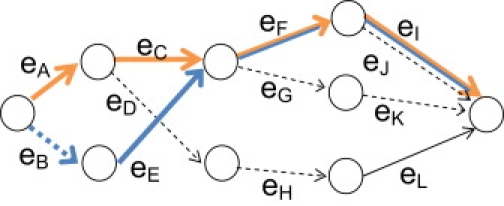
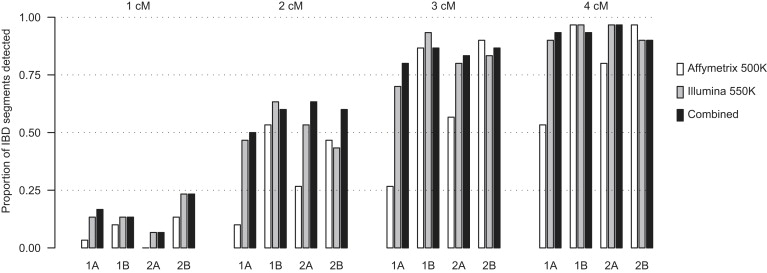
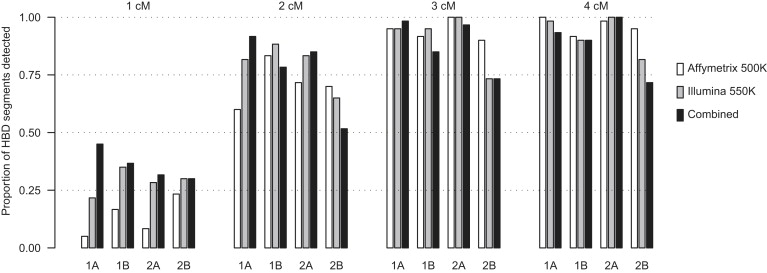
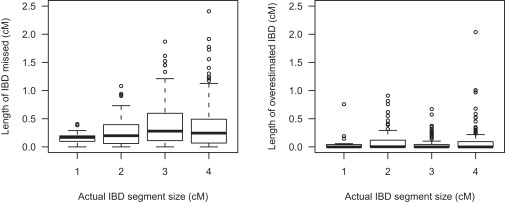
References
-
- Nelson, S., Merriman, B., Chen, Z., Ogdie, M., Stone, J., and Strom, S. (2006). Applications of Pedigree-Free Identity-By-Descent Mapping to Localizing Disease Genes [abstract 1530]. Presented at the annual meeting of The American Society of Human Genetics, October 11, 2006, New Orleans, LA, USA. Available from http://www.ashg.org/genetics/ashg06s/.
-
- Albrechtsen A., Sand Korneliussen T., Moltke I., van Overseem Hansen T., Nielsen F.C., Nielsen R. Relatedness mapping and tracts of relatedness for genome-wide data in the presence of linkage disequilibrium. Genet. Epidemiol. 2009;33:266–274. - PubMed
-
- Wijsman E.M., Amos C.I. Genetic analysis of simulated oligogenic traits in nuclear and extended pedigrees: summary of GAW10 contributions. Genet. Epidemiol. 1997;14:719–735. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
