

Helicobacter pylori CagL activates ADAM17 to induce repression of the gastric H, K-ATPase alpha subunit
- PMID: 20303353
- PMCID: PMC2902712
- DOI: 10.1053/j.gastro.2010.03.036
Helicobacter pylori CagL activates ADAM17 to induce repression of the gastric H, K-ATPase alpha subunit
Abstract
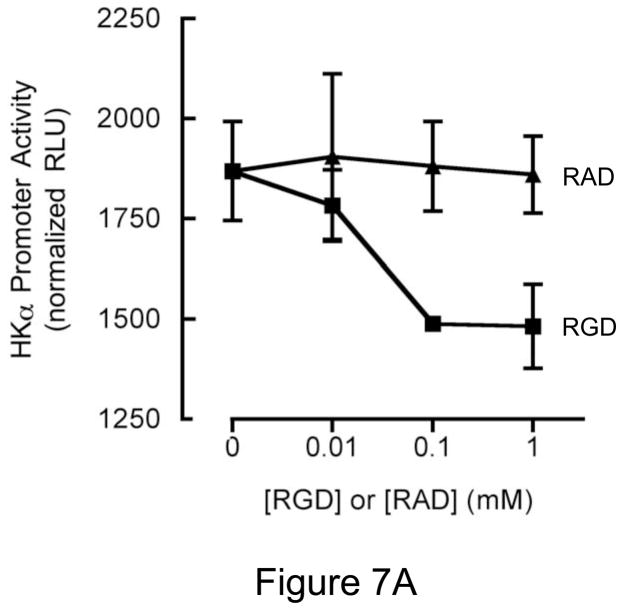
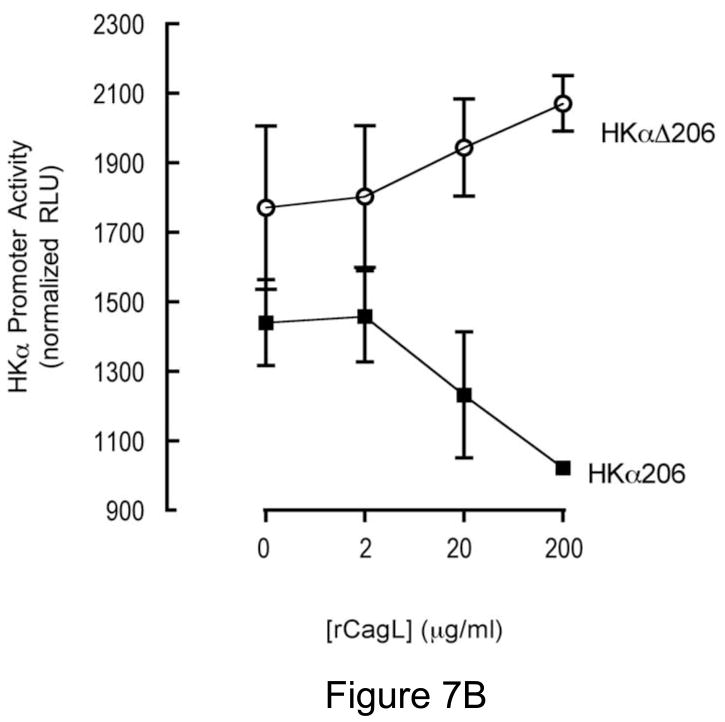
Background & aims: Infection with Helicobacter pylori represses expression of the gastric H, K-adenosine triphosphatase alpha-subunit (HKalpha), which could contribute to transient hypochlorhydria. CagL, a pilus protein component of the H pylori type IV secretion system, binds to the integrin alpha(5)beta1 to mediate translocation of virulence factors into the host cell and initiate signaling. alpha(5)beta1 binds a disintegrin and metalloprotease (ADAM) 17, a metalloenzyme that catalyzes ectodomain shedding of receptor tyrosine kinase ligands. We investigated whether H pylori-induced repression of HKalpha is mediated by CagL activation of ADAM17 and release of heparin-binding epidermal growth factor (HB-EGF).
Methods: HKalpha promoter and ADAM17 activity were measured in AGS gastric epithelial cells transfected with HKalpha promoter-reporter constructs or ADAM17-specific small interfering RNAs and infected with H pylori. HB-EGF secretion was measured by enzyme-linked immunosorbent assay analysis, and ADAM17 interaction with integrins was investigated by coimmunoprecipitation analyses.
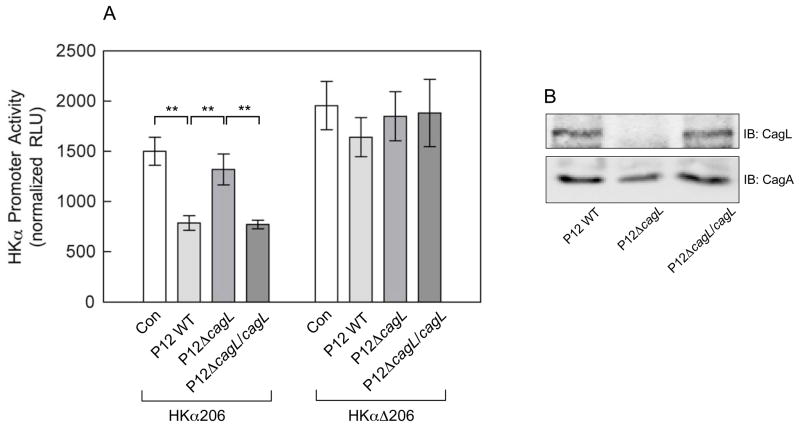
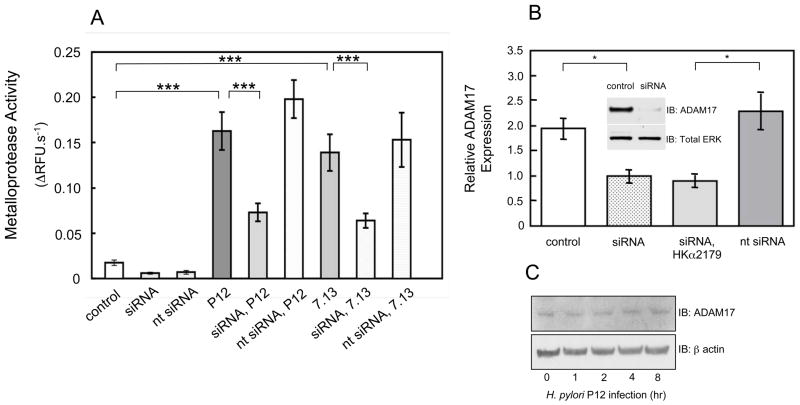
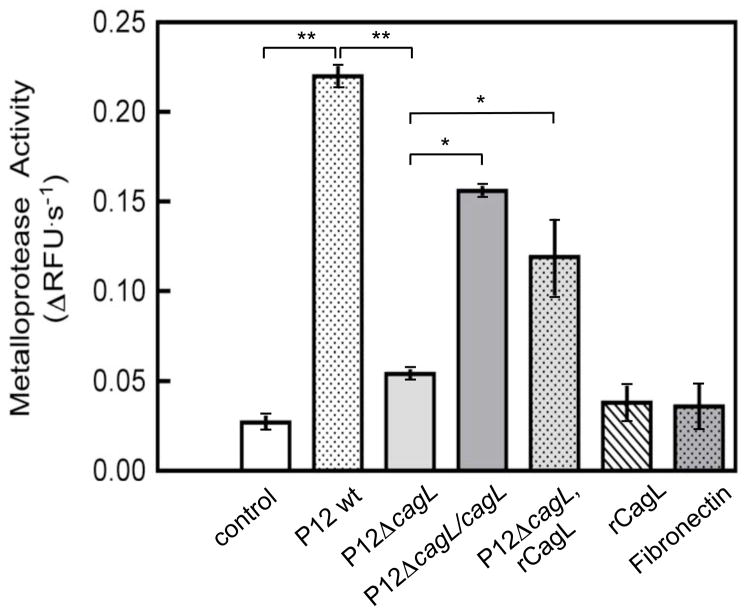
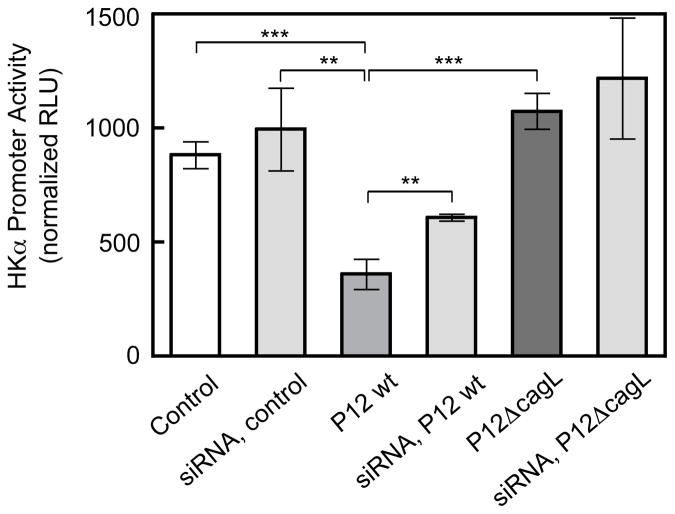
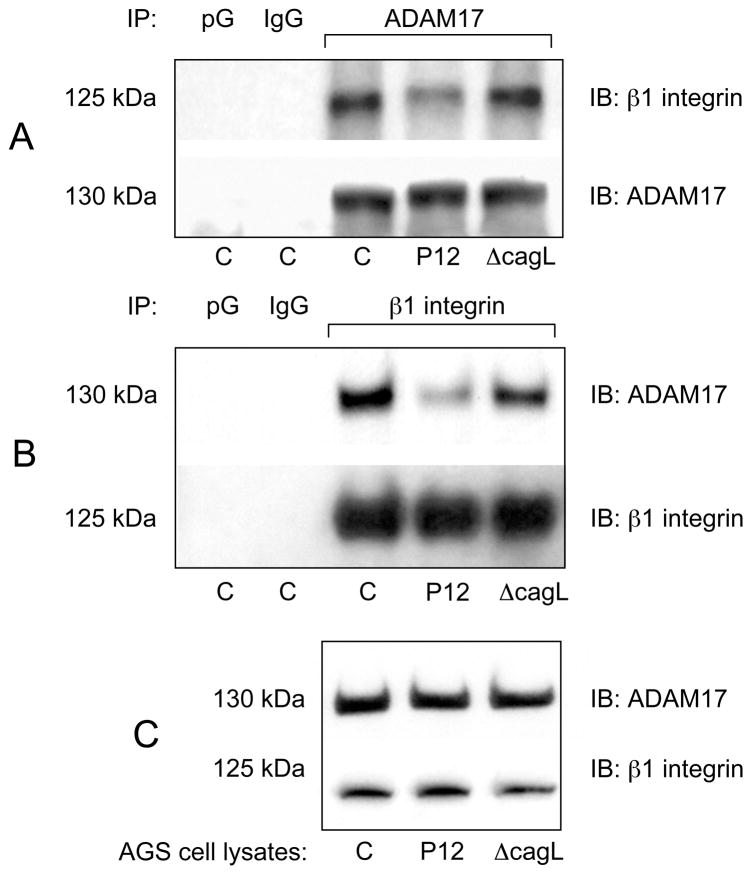
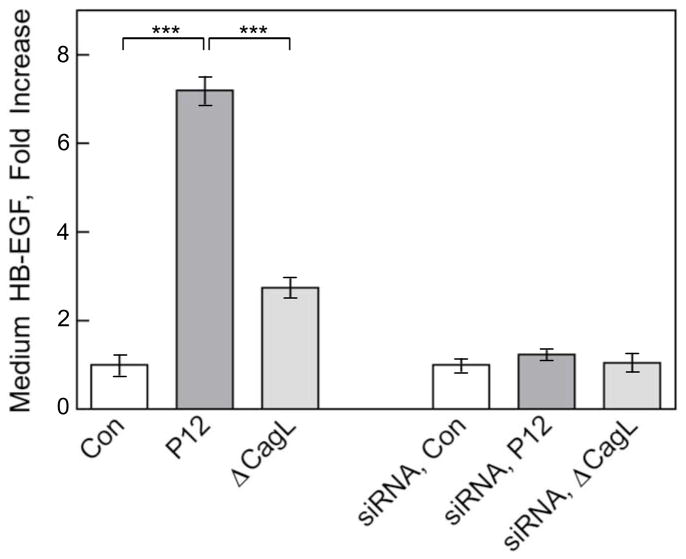
Results: Infection of AGS cells with wild-type H pylori or an H pylori cagL-deficient isogenic mutant that also contained a wild-type version of cagL (P12DeltacagL/cagL) repressed HKalpha promoter-Luc reporter activity and stimulated ADAM17 activity. Both responses were inhibited by point mutations in the nuclear factor-kappaB binding site of HKalpha or by infection with P12DeltacagL. Small interfering RNA-mediated silencing of ADAM17 in AGS cells inhibited the repression of wild-type HKalpha promoter and reduced ADAM17 activity and HB-EGF production, compared to controls. Coimmunoprecipitation studies of AGS lysates showed that wild-type H pylori disrupted ADAM17-alpha5beta1 complexes.
Conclusions: During acute H pylori infection, CagL dissociates ADAM17 from the integrin alpha(5)beta1 and activates ADAM17-dependent, nuclear factor-kappaB-mediated repression of HKalpha. This might contribute to transient hypochlorhydria in patients with H pylori infection.
Copyright 2010 AGA Institute. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Graham DY, Alpert LC, Smith JL, et al. Iatrogenic Campylobacter pylori infection is a cause of epidemic achlorhydria. Am J Gastroenterol. 1988;83:974–80. - PubMed
-
- Morris A, Nicholson G. Experimental and accidental C. pylori infection in humans. In: Blaser MJ, editor. Campylobacter pylori in Gastritis and Peptic Ulcer Disease. New York: Igaku-Shoin; 1989. pp. 61–72.
-
- Zavros Y, Eaton KA, Kang W, et al. Chronic gastritis in the hypochlorhydric gastrin-deficient mouse progresses to adenocarcinoma. Oncogene. 2005;24:2354–66. - PubMed
-
- Peek RM, Jr, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer. 2002;2:28–37. - PubMed
-
- O’Toole D, Abdel-Latif MM, Long A, et al. Low pH and Helicobacter pylori increase nuclear factor kappa B binding in gastric epithelial cells: a common pathway for epithelial cell injury? J Cell Biochem. 2005;96:589–98. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
