

DNA methylation profiling using the methylated-CpG island recovery assay (MIRA)
- PMID: 20304072
- PMCID: PMC2910839
- DOI: 10.1016/j.ymeth.2010.03.004
DNA methylation profiling using the methylated-CpG island recovery assay (MIRA)
Abstract
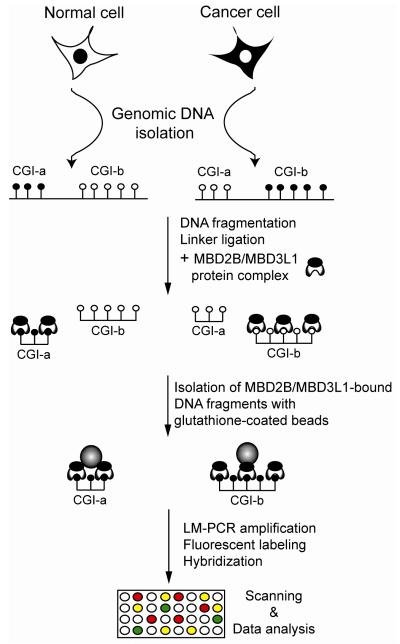
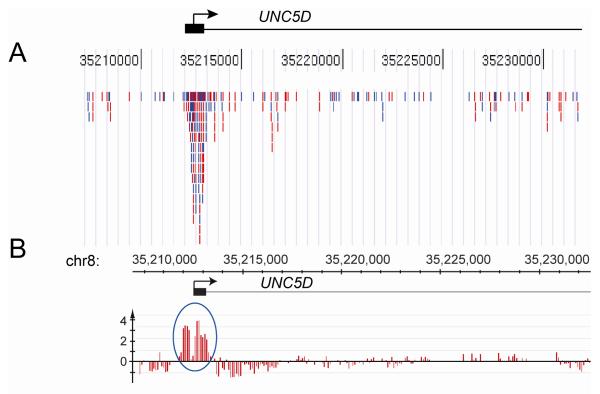
The methylated-CpG island recovery assay (MIRA) exploits the intrinsic specificity and the high affinity of a methylated-CpG-binding protein complex (MBD2B and MBD3L1) to methylated CpG dinucleotides in genomic DNA. The MIRA approach works on double-stranded DNA and does not depend on the application of methylation-sensitive restriction enzymes. It can be performed on a few hundred nanograms of genomic DNA. Recently, the MIRA technique has been used to profile DNA methylation patterns at a resolution of 100 base pairs along the entire genome of normal human B-lymphocytes. The MIRA method is compatible with microarray and next generation DNA sequencing approaches. We describe the principles and details of this method applied for methylation profiling of genomes containing methylated CpG sequences.
Copyright © 2010 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
MIRA-seq for DNA methylation analysis of CpG islands.Epigenomics. 2015 Aug;7(5):695-706. doi: 10.2217/epi.15.33. Epub 2015 Apr 17. Epigenomics. 2015. PMID: 25881900 Free PMC article.
-
Methylated-CpG Island Recovery Assay.Methods Mol Biol. 2011;791:125-33. doi: 10.1007/978-1-61779-316-5_10. Methods Mol Biol. 2011. PMID: 21913076 Free PMC article.
-
The MIRA method for DNA methylation analysis.Methods Mol Biol. 2009;507:65-75. doi: 10.1007/978-1-59745-522-0_6. Methods Mol Biol. 2009. PMID: 18987807 Free PMC article.
-
DNA methylation patterns in lung carcinomas.Semin Cancer Biol. 2009 Jun;19(3):181-7. doi: 10.1016/j.semcancer.2009.02.008. Epub 2009 Feb 20. Semin Cancer Biol. 2009. PMID: 19429482 Free PMC article. Review.
-
The methyl-CpG binding domain and the evolving role of DNA methylation in animals.Trends Genet. 2003 May;19(5):269-77. doi: 10.1016/S0168-9525(03)00080-5. Trends Genet. 2003. PMID: 12711219 Review.
Cited by
-
Chromosome-wide analysis of parental allele-specific chromatin and DNA methylation.Mol Cell Biol. 2011 Apr;31(8):1757-70. doi: 10.1128/MCB.00961-10. Epub 2011 Feb 14. Mol Cell Biol. 2011. PMID: 21321082 Free PMC article.
-
High Resolution Imaging of DNA Methylation Dynamics using a Zebrafish Reporter.Sci Rep. 2017 Jul 14;7(1):5430. doi: 10.1038/s41598-017-05648-8. Sci Rep. 2017. PMID: 28710355 Free PMC article.
-
DNA methylation biomarkers for lung cancer.Tumour Biol. 2012 Apr;33(2):287-96. doi: 10.1007/s13277-011-0282-2. Epub 2011 Dec 6. Tumour Biol. 2012. PMID: 22143938
-
Methods for high-throughput MethylCap-Seq data analysis.BMC Genomics. 2012;13 Suppl 6(Suppl 6):S14. doi: 10.1186/1471-2164-13-S6-S14. Epub 2012 Oct 26. BMC Genomics. 2012. PMID: 23134780 Free PMC article.
-
Genomewide analyses of pathogenic and regulatory T cells of NOD mice reveal a significant difference in DNA methylation on chromosome X.J Genet. 2016 Dec;95(4):1023-1029. doi: 10.1007/s12041-016-0729-8. J Genet. 2016. PMID: 27994204 No abstract available.
References
-
- Rauch TA, Pfeifer GP. Methods for assessing genome-wide DNA methylation. In: Tollefsbol T, editor. Handbook of Epigenetics: The New Molecular and Medical Genetics. Elsevier; 2010. in press.
-
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, Edsall L, Antosiewicz-Bourget J, Stewart R, Ruotti V, Millar AH, Thomson JA, Ren B, Ecker JR. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. - PMC - PubMed
-
- Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, Burton J, Cox TV, Davies R, Down TA, Haefliger C, Horton R, Howe K, Jackson DK, Kunde J, Koenig C, Liddle J, Niblett D, Otto T, Pettett R, Seemann S, Thompson C, West T, Rogers J, Olek A, Berlin K, Beck S. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet. 2006;38:1378–1385. - PMC - PubMed
-
- Rauch T, Li H, Wu X, Pfeifer GP. MIRA-assisted microarray analysis, a new technology for the determination of DNA methylation patterns, identifies frequent methylation of homeodomain-containing genes in lung cancer cells. Cancer Res. 2006;66:7939–7947. - PubMed
-
- Weber M, Davies JJ, Wittig D, Oakeley EJ, Haase M, Lam WL, Schubeler D. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet. 2005;37:853–862. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
