

Synthesis and pharmacological characterization of a new series of 5,7-disubstituted-[1,2,4]triazolo[1,5-a][1,3,5]triazine derivatives as adenosine receptor antagonists: A preliminary inspection of ligand-receptor recognition process
- PMID: 20304654
- PMCID: PMC3106415
- DOI: 10.1016/j.bmc.2010.02.039
Synthesis and pharmacological characterization of a new series of 5,7-disubstituted-[1,2,4]triazolo[1,5-a][1,3,5]triazine derivatives as adenosine receptor antagonists: A preliminary inspection of ligand-receptor recognition process
Abstract
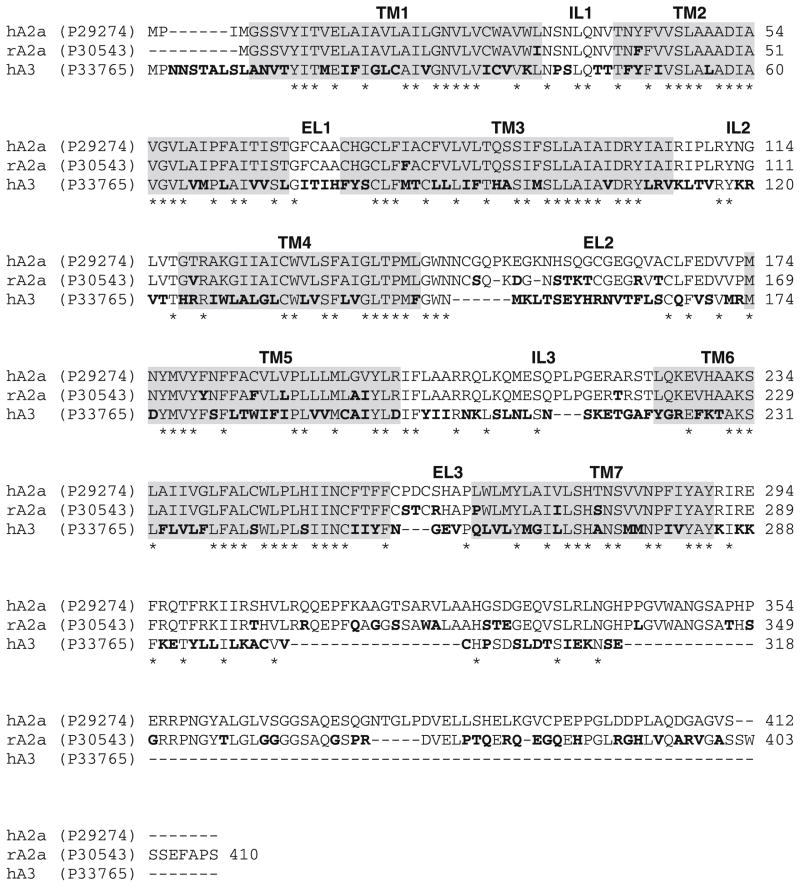
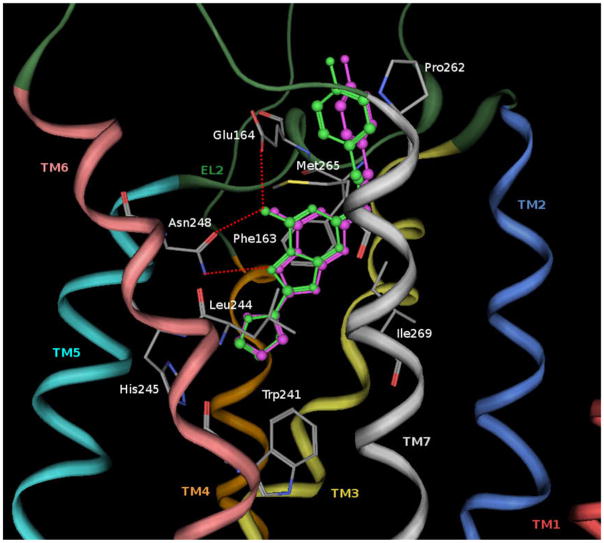
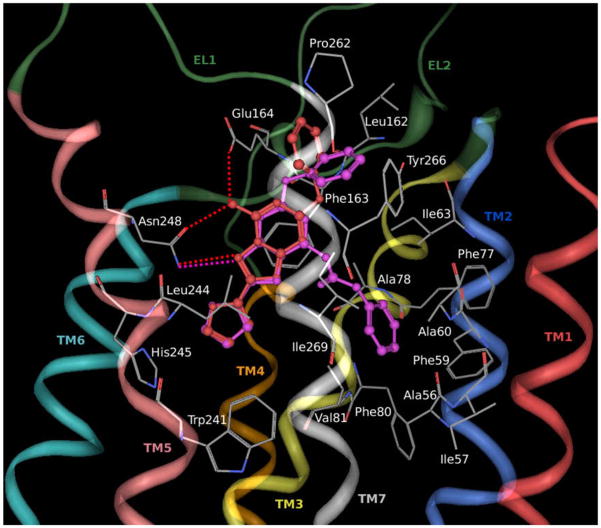
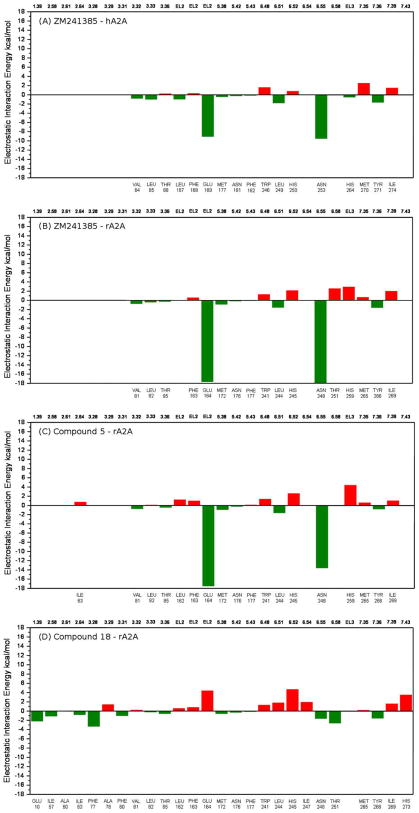
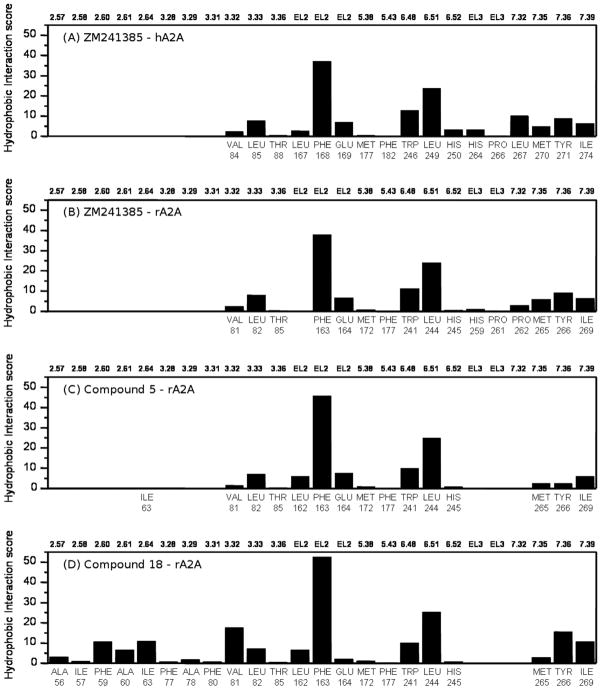
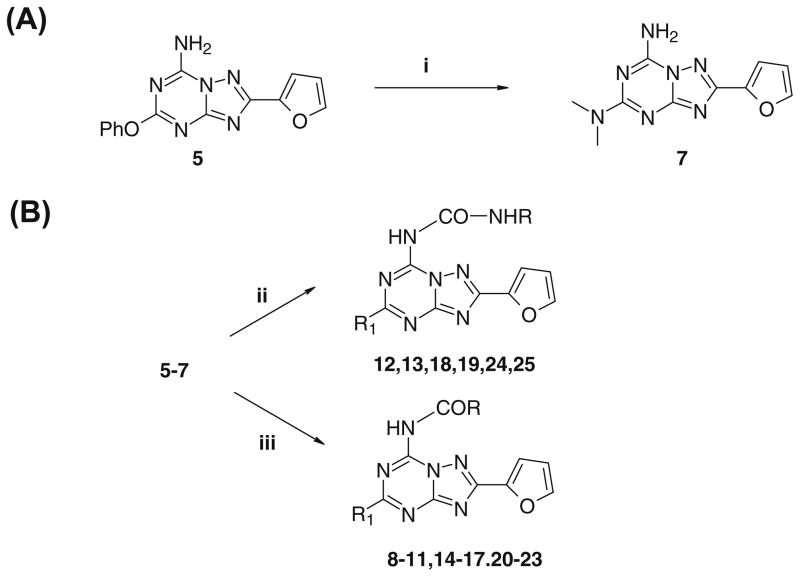
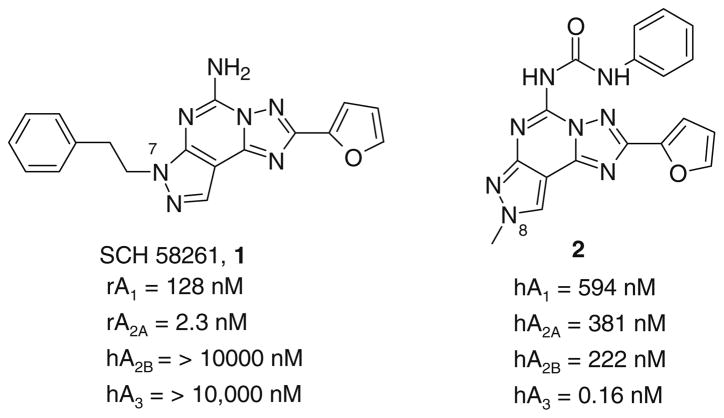
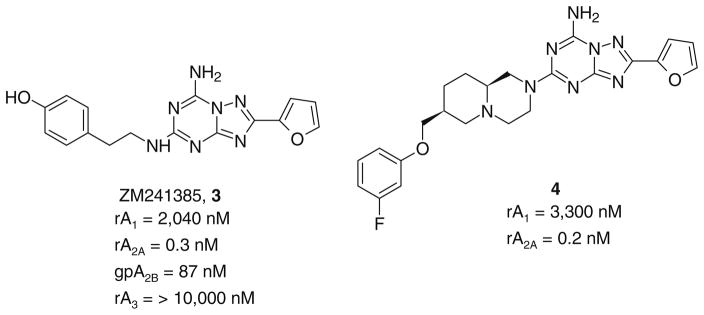
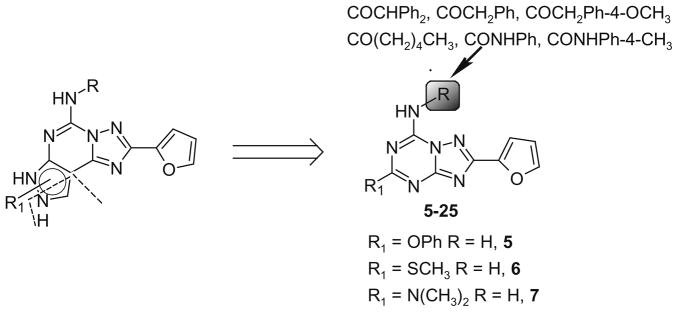
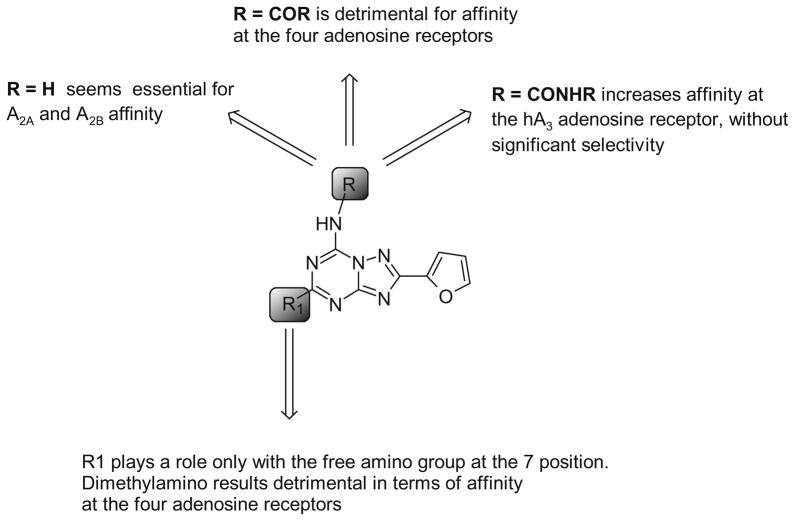
A new series of triazolotriazines variously substituted at the C5 and N7 (5-25) positions was synthesized and fully characterized at the four adenosine receptor (AR) subtypes. In particular, arylacetyl or arylcarbamoyl moieties were introduced at the N7 position, which enhanced affinity at the hA(2B) and hA(3) ARs, respectively, when utilized on the pyrazolo-triazolopyrimidine nucleus as we reported in the past. In general, compounds with a free amino group at the 7 position (5, 6), showed good affinity at the rat (r) A(2A) AR (range 18.3-96.5nM), while the introduction of a phenylcarbamoyl moiety at the N7 position (12, 19, 24) slightly increased the affinity at the hA(3) AR (range 311-633nM) with respect to the unsubstituted derivatives. The binding profiles of the synthesized analogues seemed to correlate with the substitutions at the C5 and N7 positions. At the hA(2B) AR, derivative 5, which contained a free amino group at the 7 position, was the most potent (EC(50) 3.42microM) and could represent a starting point for searching new non-xanthine hA(2B) AR antagonists. Molecular models of the rA(2A) and hA(3) ARs were constructed by homology to the recently reported crystallographic structure of the hA(2A) AR. A preliminary receptor-driven structure-activity relationship (SAR) based on the analysis of antagonist docking has been provided.
Copyright 2010 Elsevier Ltd. All rights reserved.
Figures
Similar articles
-
Synthesis and biological evaluation of a new series of 1,2,4-triazolo[1,5-a]-1,3,5-triazines as human A(2A) adenosine receptor antagonists with improved water solubility.J Med Chem. 2011 Feb 10;54(3):877-89. doi: 10.1021/jm101349u. Epub 2011 Jan 7. J Med Chem. 2011. PMID: 21214204 Free PMC article.
-
Synthesis, biological activity, and molecular modeling investigation of new pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine derivatives as human A(3) adenosine receptor antagonists.J Med Chem. 2002 Feb 14;45(4):770-80. doi: 10.1021/jm0109614. J Med Chem. 2002. PMID: 11831890
-
5,7-Disubstituted-[1,2,4]triazolo[1,5-a][1,3,5]triazines as pharmacological tools to explore the antagonist selectivity profiles toward adenosine receptors.Eur J Med Chem. 2016 Jan 27;108:529-541. doi: 10.1016/j.ejmech.2015.12.019. Epub 2015 Dec 15. Eur J Med Chem. 2016. PMID: 26717203
-
Potent adenosine A1 and A2A receptors antagonists: recent developments.Curr Med Chem. 2006;13(30):3609-25. doi: 10.2174/092986706779026093. Curr Med Chem. 2006. PMID: 17168726 Review.
-
Introduction to adenosine receptors as therapeutic targets.Handb Exp Pharmacol. 2009;(193):1-24. doi: 10.1007/978-3-540-89615-9_1. Handb Exp Pharmacol. 2009. PMID: 19639277 Free PMC article. Review.
Cited by
-
Synthesis and biological evaluation of a new series of 1,2,4-triazolo[1,5-a]-1,3,5-triazines as human A(2A) adenosine receptor antagonists with improved water solubility.J Med Chem. 2011 Feb 10;54(3):877-89. doi: 10.1021/jm101349u. Epub 2011 Jan 7. J Med Chem. 2011. PMID: 21214204 Free PMC article.
-
Progress in structure based drug design for G protein-coupled receptors.J Med Chem. 2011 Jul 14;54(13):4283-311. doi: 10.1021/jm200371q. Epub 2011 Jun 15. J Med Chem. 2011. PMID: 21615150 Free PMC article. Review. No abstract available.
-
A Pharmacological Overview and Recent Patent of Triazine Scaffold in Drug Development: A Review.Curr Org Synth. 2025;22(3):310-327. doi: 10.2174/0115701794272212240307092318. Curr Org Synth. 2025. PMID: 40259585 Review.
-
Recent developments in adenosine receptor ligands and their potential as novel drugs.Biochim Biophys Acta. 2011 May;1808(5):1290-308. doi: 10.1016/j.bbamem.2010.12.017. Epub 2010 Dec 23. Biochim Biophys Acta. 2011. PMID: 21185259 Free PMC article. Review.
-
Expression of adenosine receptors in monocytes from patients with bronchial asthma.Biochem Biophys Res Commun. 2015 Sep 4;464(4):1314-1320. doi: 10.1016/j.bbrc.2015.07.141. Epub 2015 Jul 30. Biochem Biophys Res Commun. 2015. PMID: 26232643 Free PMC article.
References
-
- Downey JM, Cohen MV, Ytrehus K, Liu Y. Ann NY Acad Sci. 1994;723:82. - PubMed
-
- Auchampach JA, Jin X, Wan TC, Caughey GH, Linden J. Mol Pharmacol. 1997;52:846. - PubMed
-
- Fredholm BB. Int Rev Neurobiol. 1997;40:259. - PubMed
-
- Feoktistov I, Polosa R, Holgate ST, Biaggioni I. Trends Pharmacol Sci. 1998;19:148. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Chemical Information
Research Materials
Miscellaneous
