

Role of SMAD and non-SMAD signals in the development of Th17 and regulatory T cells
- PMID: 20304828
- PMCID: PMC3087811
- DOI: 10.4049/jimmunol.0903418
Role of SMAD and non-SMAD signals in the development of Th17 and regulatory T cells
Abstract
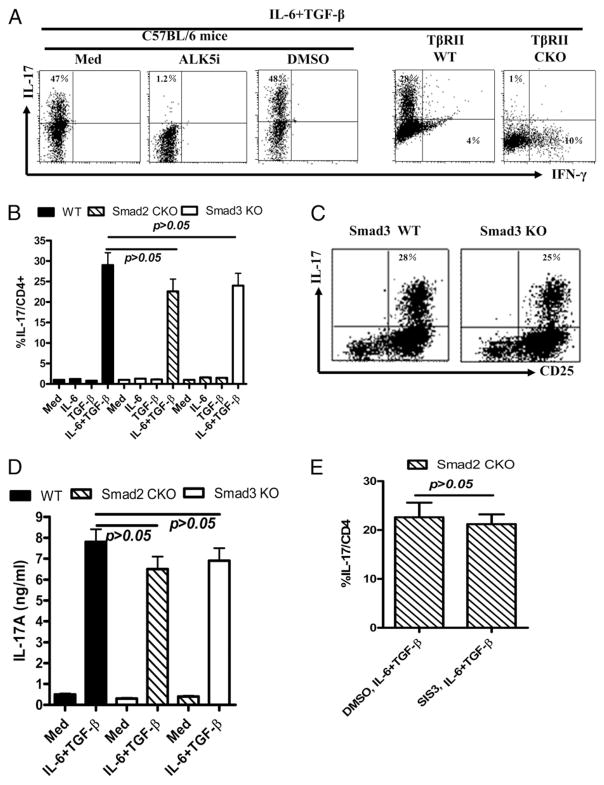
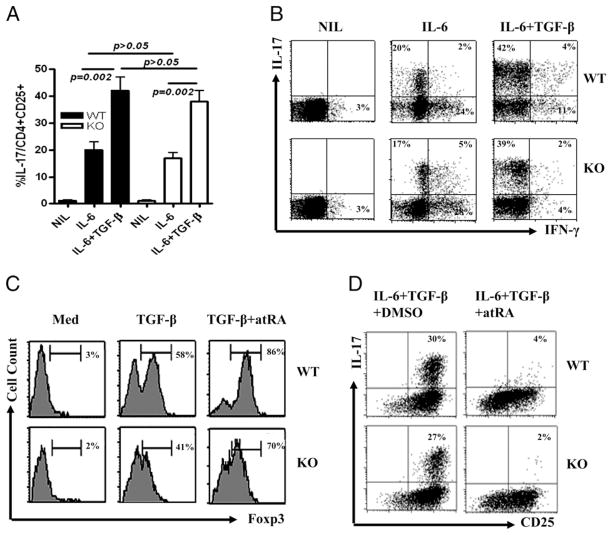
Whereas TGF-beta is essential for the development of peripherally induced Foxp3(+) regulatory T cells (iTreg cells) and Th17 cells, the intracellular signaling mechanism by which TGF-beta regulates development of both cell subsets is less understood. In this study, we report that neither Smad2 nor Smad3 gene deficiency abrogates TGF-beta-dependent iTreg induction by a deacetylase inhibitor trichostatin A in vivo, although the loss of the Smad2 or Smad3 gene partially reduces iTreg induction in vitro. Similarly, SMAD2 and SMAD3 have a redundant role in development of Th17 in vitro and in experimental autoimmune encephalomyelitis. In addition, ERK and/or JNK pathways were shown to be involved in regulating iTreg cells, whereas the p38 pathway predominately modulated Th17 and experimental autoimmune encephalomyelitis induction. Therefore, selective targeting of these intracellular TGF-beta signaling pathways during iTreg and Th17 cell development might lead to the development of therapies in treating autoimmune and other chronic inflammatory diseases.
Conflict of interest statement
The authors have no financial conflicts of interest.
Figures






Similar articles
-
Smad2 and Smad3 are redundantly essential for the TGF-beta-mediated regulation of regulatory T plasticity and Th1 development.J Immunol. 2010 Jul 15;185(2):842-55. doi: 10.4049/jimmunol.0904100. Epub 2010 Jun 14. J Immunol. 2010. PMID: 20548029
-
Retinoic acid increases Foxp3+ regulatory T cells and inhibits development of Th17 cells by enhancing TGF-beta-driven Smad3 signaling and inhibiting IL-6 and IL-23 receptor expression.J Immunol. 2008 Aug 15;181(4):2277-84. doi: 10.4049/jimmunol.181.4.2277. J Immunol. 2008. PMID: 18684916 Free PMC article.
-
Smad2 positively regulates the generation of Th17 cells.J Biol Chem. 2010 Sep 17;285(38):29039-43. doi: 10.1074/jbc.C110.155820. Epub 2010 Jul 28. J Biol Chem. 2010. PMID: 20667820 Free PMC article.
-
The dynamics of effector T cells and Foxp3+ regulatory T cells in the promotion and regulation of autoimmune encephalomyelitis.J Neuroimmunol. 2007 Nov;191(1-2):51-60. doi: 10.1016/j.jneuroim.2007.09.009. Epub 2007 Oct 3. J Neuroimmunol. 2007. PMID: 17916388 Free PMC article. Review.
-
TGF-β function in immune suppression.Curr Top Microbiol Immunol. 2011;350:127-47. doi: 10.1007/82_2010_87. Curr Top Microbiol Immunol. 2011. PMID: 20680806 Review.
Cited by
-
Selective Impairment of TH17-Differentiation and Protection against Autoimmune Arthritis after Overexpression of BCL2A1 in T Lymphocytes.PLoS One. 2016 Jul 19;11(7):e0159714. doi: 10.1371/journal.pone.0159714. eCollection 2016. PLoS One. 2016. PMID: 27433938 Free PMC article.
-
Regulatory T cells vs Th17: differentiation of Th17 versus Treg, are the mutually exclusive?Am J Clin Exp Immunol. 2013 Feb 27;2(1):94-106. Print 2013. Am J Clin Exp Immunol. 2013. PMID: 23885327 Free PMC article.
-
Regulators involved in trophoblast syncytialization in the placenta of intrauterine growth restriction.Front Endocrinol (Lausanne). 2023 Jan 31;14:1107182. doi: 10.3389/fendo.2023.1107182. eCollection 2023. Front Endocrinol (Lausanne). 2023. PMID: 36798658 Free PMC article. Review.
-
RORγt phosphorylation protects against T cell-mediated inflammation.Cell Rep. 2022 Mar 15;38(11):110520. doi: 10.1016/j.celrep.2022.110520. Cell Rep. 2022. PMID: 35294872 Free PMC article.
-
TGF-β-induced CD4+Foxp3+ T cells attenuate acute graft-versus-host disease by suppressing expansion and killing of effector CD8+ cells.J Immunol. 2014 Oct 1;193(7):3388-97. doi: 10.4049/jimmunol.1400207. Epub 2014 Aug 25. J Immunol. 2014. PMID: 25156367 Free PMC article.
References
-
- Zheng SG, Wang J, Wang P, Gray JD, Horwitz DA. IL-2 is essential for TGF-beta to convert naive CD4+CD25− cells to CD25+Foxp3+ regulatory T cells and for expansion of these cells. J Immunol. 2007;178:2018–2027. - PubMed
-
- Davidson TS, DiPaolo RJ, Andersson J, Shevach EM. Cutting Edge: IL-2 is essential for TGF-beta-mediated induction of Foxp3+ T regulatory cells. J Immunol. 2007;178:4022–4026. - PubMed
-
- Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. - PubMed
-
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. - PubMed
-
- Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
