

Transcription-dependent activation of ataxia telangiectasia mutated prevents DNA-dependent protein kinase-mediated cell death in response to topoisomerase I poison
- PMID: 20304914
- PMCID: PMC2865312
- DOI: 10.1074/jbc.M110.101808
Transcription-dependent activation of ataxia telangiectasia mutated prevents DNA-dependent protein kinase-mediated cell death in response to topoisomerase I poison
Abstract
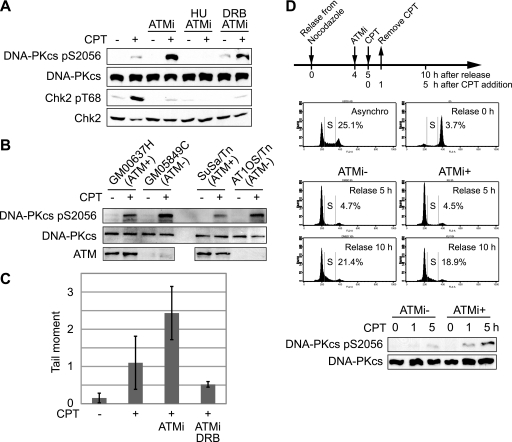
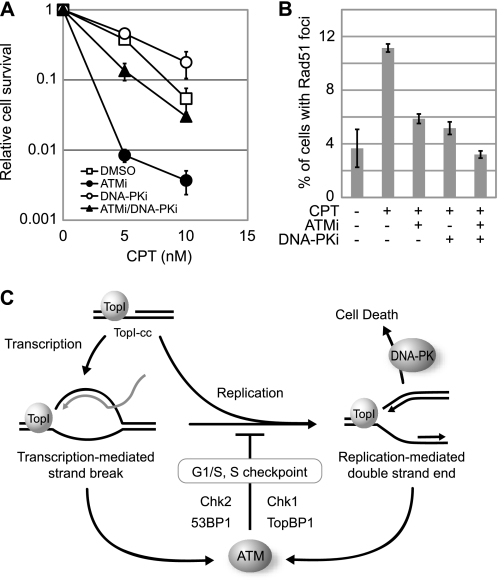
Camptothecin (CPT) is a topoisomerase I inhibitor, derivatives of which are being used for cancer chemotherapy. CPT-induced DNA double-strand breaks (DSBs) are considered a major cause of its tumoricidal activity, and it has been shown that CPT induces DNA damage signaling through the phosphatidylinositol 3-kinase-related kinases, including ATM (ataxia telangiectasia mutated), ATR (ATM and Rad3-related), and DNA-PK (DNA-dependent protein kinase). In addition, CPT causes DNA strand breaks mediated by transcription, although the downstream signaling events are less well characterized. In this study, we show that CPT-induced activation of ATM requires transcription. Mechanistically, transcription inhibition suppressed CPT-dependent activation of ATM and blocked recruitment of the DNA damage mediator p53-binding protein 1 (53BP1) to DNA damage sites, whereas ATM inhibition abrogated CPT-induced G(1)/S and S phase checkpoints. Functional inactivation of ATM resulted in DNA replication-dependent hyperactivation of DNA-PK in CPT-treated cells and dramatic CPT hypersensitivity. On the other hand, simultaneous inhibition of ATM and DNA-PK partially restored CPT resistance, suggesting that activation of DNA-PK is proapoptotic in the absence of ATM. Correspondingly, comet assay and cell cycle synchronization experiments suggested that transcription collapse occurring as the result of CPT treatment are converted to frank double-strand breaks when ATM-deficient cells bypass the G(1)/S checkpoint. Thus, ATM suppresses DNA-PK-dependent cell death in response to topoisomerase poisons, a finding with potential clinical implications.
Figures
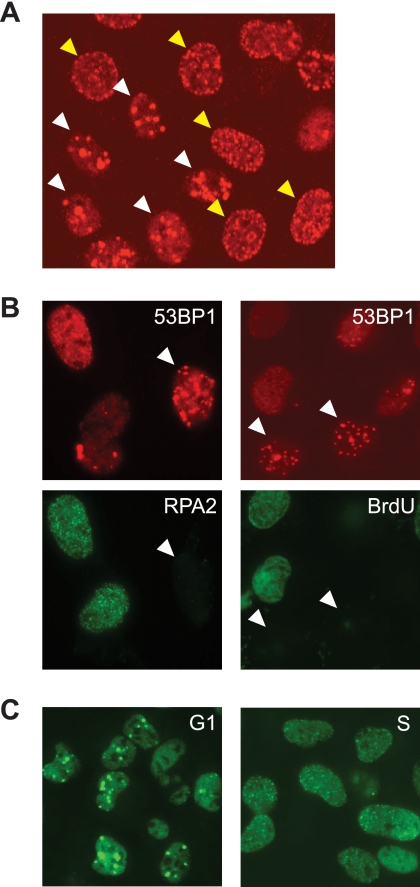
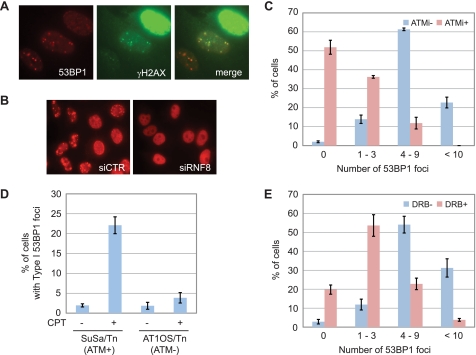
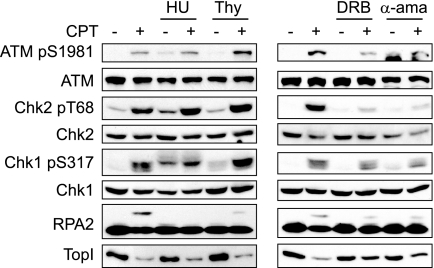
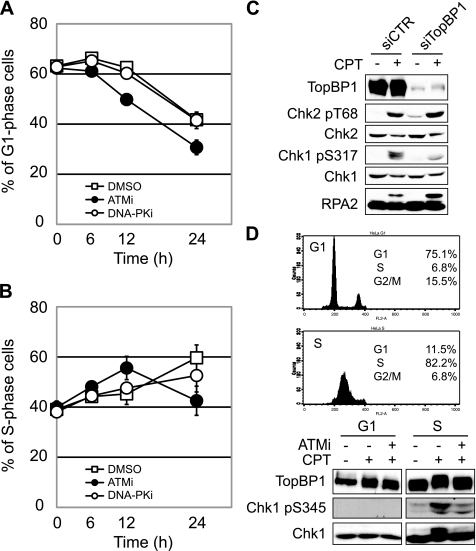
References
-
- Pommier Y. (2006) Nat. Rev. Cancer 6, 789–802 - PubMed
-
- Arnaudeau C., Lundin C., Helleday T. (2001) J. Mol. Biol. 307, 1235–1245 - PubMed
-
- Klein H. L., Kreuzer K. N. (2002) Mol. Cell 9, 471–480 - PubMed
-
- Cliby W. A., Lewis K. A., Lilly K. K., Kaufmann S. H. (2002) J. Biol. Chem. 277, 1599–1606 - PubMed
-
- Wang J. L., Wang X., Wang H., Iliakis G., Wang Y. (2002) Cell Cycle 1, 267–272 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
