

Defective nucleolar localization and dominant interfering properties of a parafibromin L95P missense mutant causing the hyperparathyroidism-jaw tumor syndrome
- PMID: 20304979
- PMCID: PMC3098453
- DOI: 10.1677/ERC-09-0272
Defective nucleolar localization and dominant interfering properties of a parafibromin L95P missense mutant causing the hyperparathyroidism-jaw tumor syndrome
Abstract
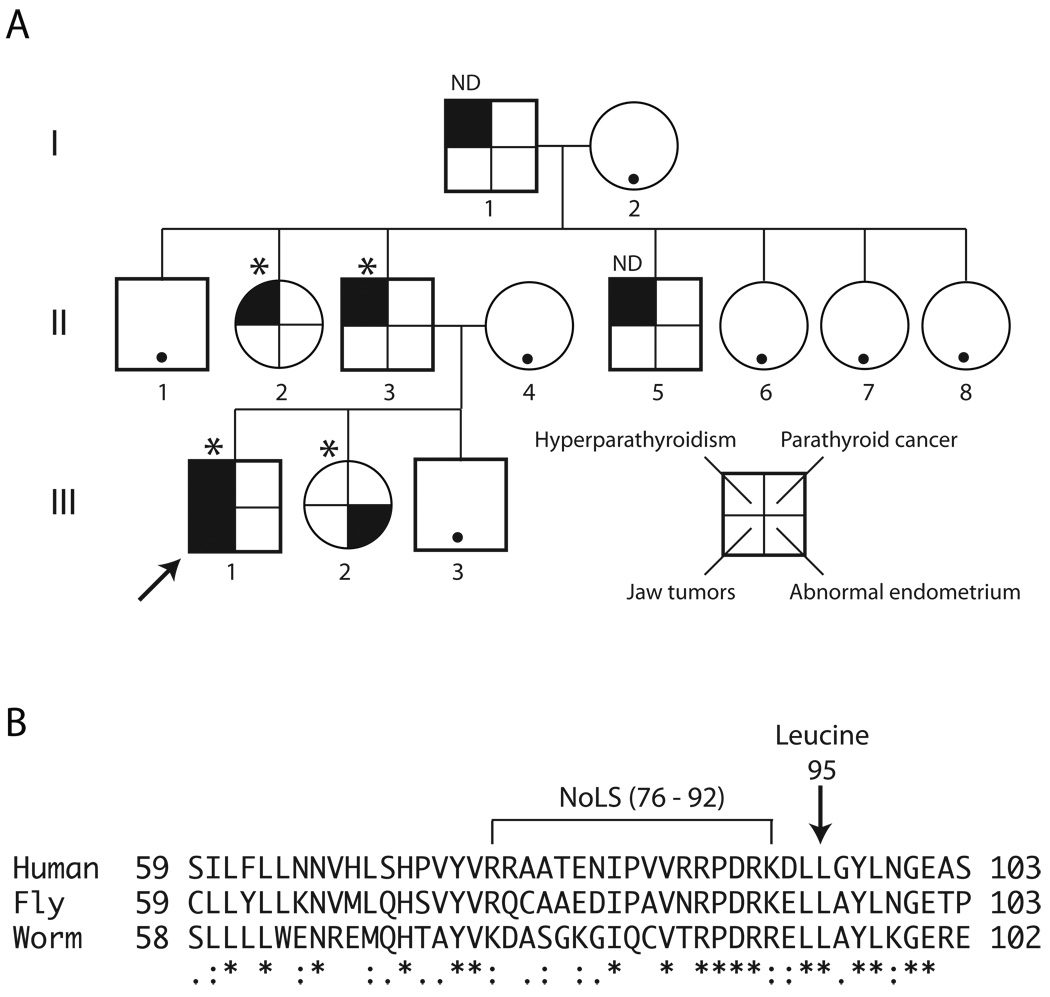
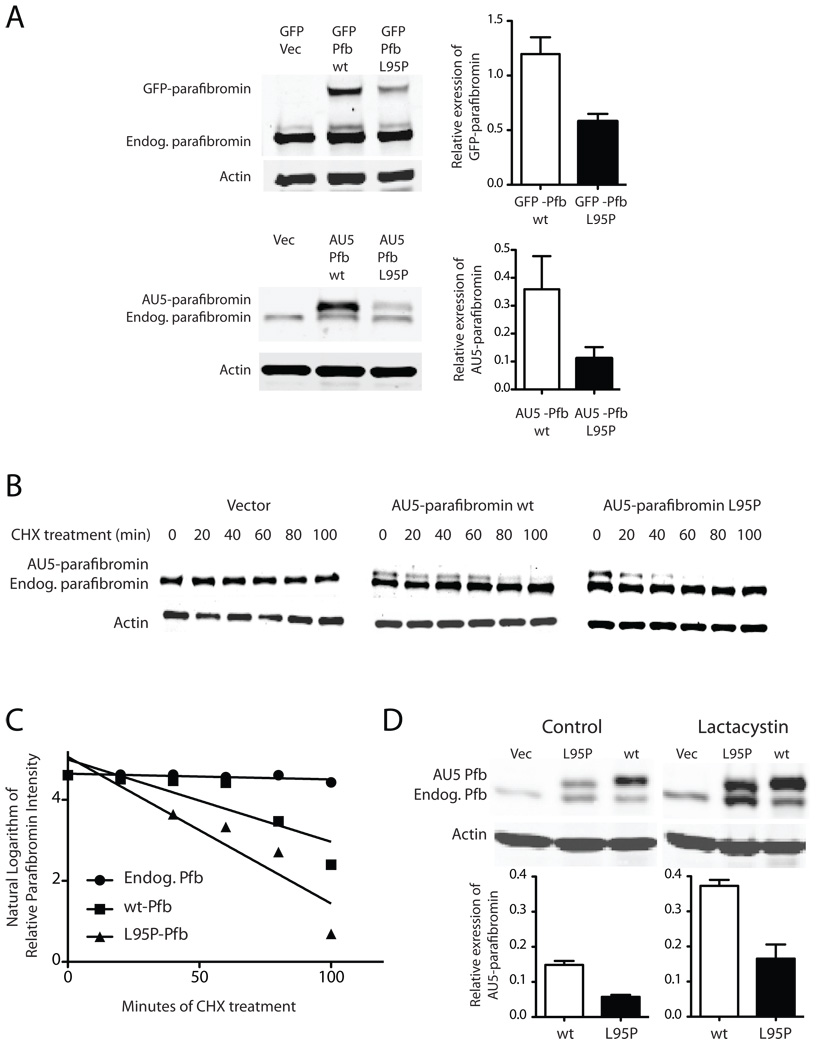
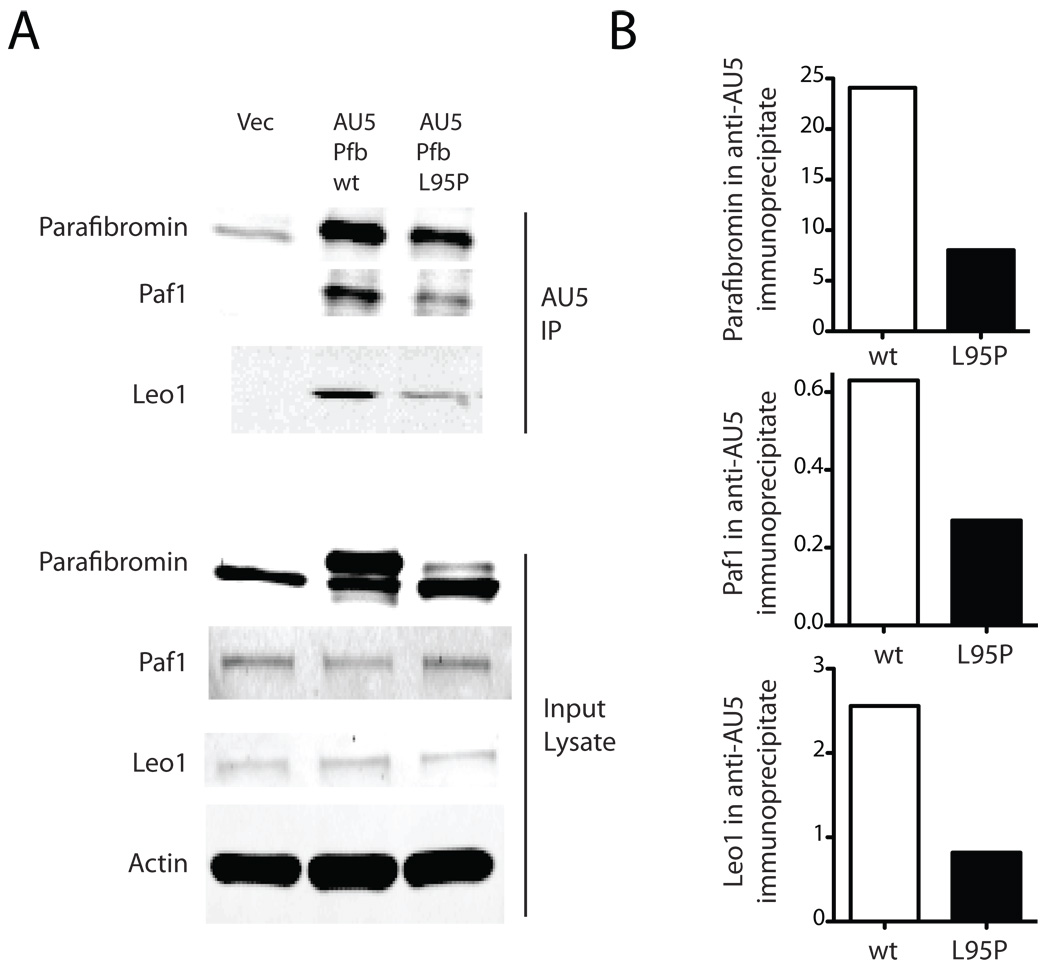
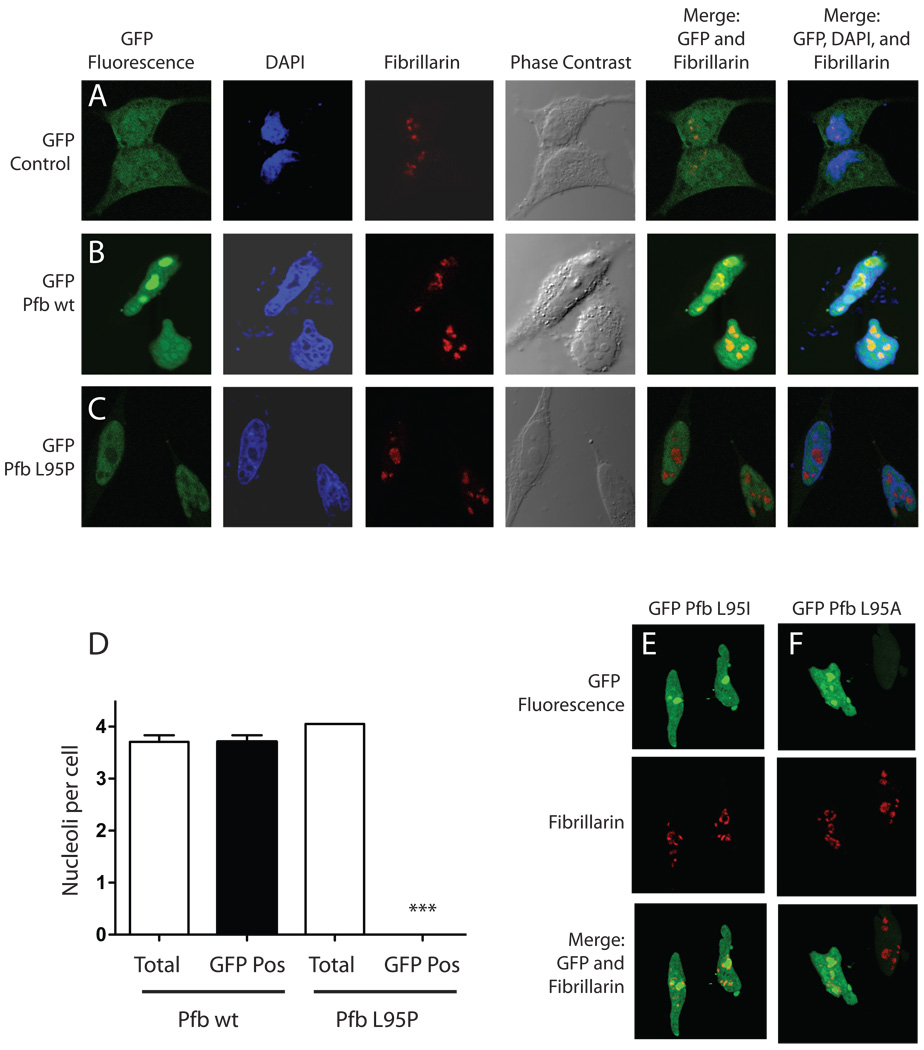
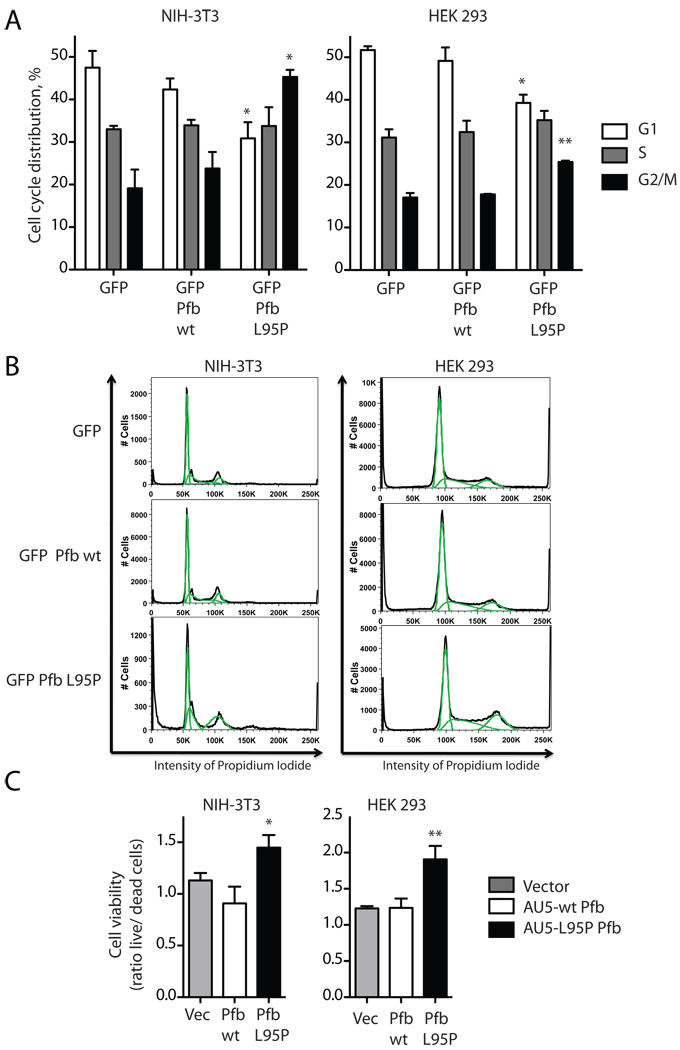
The hyperparathyroidism-jaw tumor syndrome (HPT-JT) is a familial cancer syndrome that can result from germline inactivation of HRPT2/CDC73, a putative tumor suppressor gene that encodes parafibromin, a component of the transcriptional regulatory PAF1 complex with homology to the yeast protein Cdc73p. The vast majority of HRPT2/CDC73 germline mutations identified have been truncation or frameshift mutations, and loss of function due to missense mutation is rare. We report here a kindred with HPT-JT due to a germline L95P missense mutation in parafibromin. The mutant parafibromin was studied in vitro to understand the basis of its presumed loss-of-function. When transfected in cultured cells, the L95P mutant was expressed to a lower level than wild-type (wt) parafibromin, a difference that was not overcome by inhibition of the proteasomal degradation pathway. The L95P mutant parafibromin retained the ability to assemble with endogenous PAF1 complex components as evidenced by co-immunoprecipitation. Analysis of subcellular localization showed that the L95P mutant was markedly deficient in nucleolar localization compared to the wt, an impairment likely resulting from disruption of a putative nucleolar localization signal immediately upstream of the L95P mutation. Transfection of the L95P parafibromin mutant, but not the wt, enhanced cell cycle progression and increased cell survival in NIH-3T3 and HEK 293 cells, resulting apparently from dominant interference with endogenous parafibromin action. The simultaneous loss of nucleolar localization and acquisition of a growth stimulatory phenotype with the L95P mutation raise the possibility that parafibromin must interact with targets in the nucleolus to fully execute its tumor suppressor functions.
Conflict of interest statement
None of the authors has a conflict of interest that could be perceived as prejudicing the impartiality of the research reported herein.
Figures
References
-
- Bradley KJ, Cavaco BM, Bowl MR, Harding B, Cranston T, Fratter C, Besser GM, Conceicao Pereira M, Davie MW, Dudley N, et al. Parafibromin mutations in hereditary hyperparathyroidism syndromes and parathyroid tumours. Clin Endocrinol (Oxf) 2006;64:299–306. - PubMed
-
- Bradley KJ, Hobbs MR, Buley ID, Carpten JD, Cavaco BM, Fares JE, Laidler P, Manek S, Robbins CM, Salti IS, et al. Uterine tumours are a phenotypic manifestation of the hyperparathyroidism-jaw tumour syndrome. J Intern Med. 2005;257:18–26. - PubMed
-
- Carpten JD, Robbins CM, Villablanca A, Forsberg L, Presciuttini S, Bailey-Wilson J, Simonds WF, Gillanders EM, Kennedy AM, Chen JD, et al. HRPT2, encoding parafibromin, is mutated in hyperparathyroidism-jaw tumor syndrome. Nat Genet. 2002;32:676–680. - PubMed
-
- Cetani F, Pardi E, Borsari S, Viacava P, Dipollina G, Cianferotti L, Ambrogini E, Gazzerro E, Colussi G, Berti P, et al. Genetic analyses of the HRPT2 gene in primary hyperparathyroidism: germline and somatic mutations in familial and sporadic parathyroid tumors. J Clin. Endocrinol. Metab. 2004;89:5583–5591. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
