ProBiS algorithm for detection of structurally similar protein binding sites by local structural alignment
- PMID: 20305268
- PMCID: PMC2859123
- DOI: 10.1093/bioinformatics/btq100
ProBiS algorithm for detection of structurally similar protein binding sites by local structural alignment
Abstract
Motivation: Exploitation of locally similar 3D patterns of physicochemical properties on the surface of a protein for detection of binding sites that may lack sequence and global structural conservation.
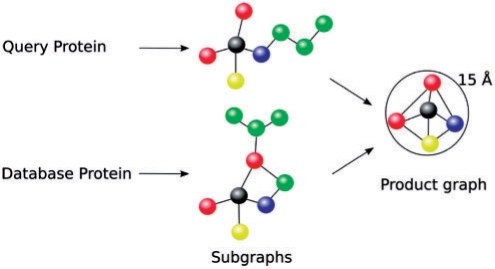
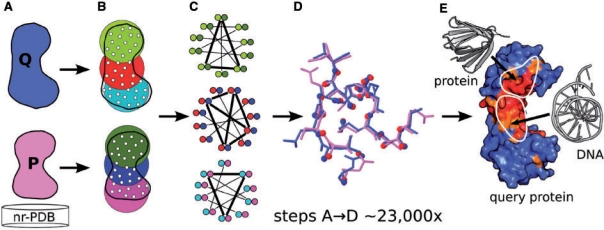
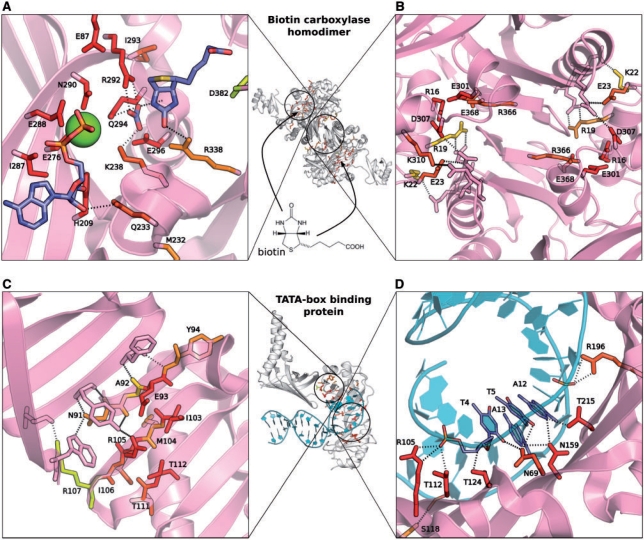
Results: An algorithm, ProBiS is described that detects structurally similar sites on protein surfaces by local surface structure alignment. It compares the query protein to members of a database of protein 3D structures and detects with sub-residue precision, structurally similar sites as patterns of physicochemical properties on the protein surface. Using an efficient maximum clique algorithm, the program identifies proteins that share local structural similarities with the query protein and generates structure-based alignments of these proteins with the query. Structural similarity scores are calculated for the query protein's surface residues, and are expressed as different colors on the query protein surface. The algorithm has been used successfully for the detection of protein-protein, protein-small ligand and protein-DNA binding sites.
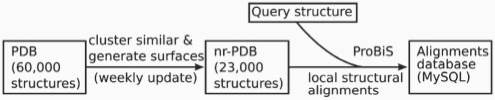
Availability: The software is available, as a web tool, free of charge for academic users at http://probis.cmm.ki.si.
Figures