

Apples and oranges: avoiding different priors in Bayesian DNA sequence analysis
- PMID: 20307305
- PMCID: PMC2859755
- DOI: 10.1186/1471-2105-11-149
Apples and oranges: avoiding different priors in Bayesian DNA sequence analysis
Abstract
Background: One of the challenges of bioinformatics remains the recognition of short signal sequences in genomic DNA such as donor or acceptor splice sites, splicing enhancers or silencers, translation initiation sites, transcription start sites, transcription factor binding sites, nucleosome binding sites, miRNA binding sites, or insulator binding sites. During the last decade, a wealth of algorithms for the recognition of such DNA sequences has been developed and compared with the goal of improving their performance and to deepen our understanding of the underlying cellular processes. Most of these algorithms are based on statistical models belonging to the family of Markov random fields such as position weight matrix models, weight array matrix models, Markov models of higher order, or moral Bayesian networks. While in many comparative studies different learning principles or different statistical models have been compared, the influence of choosing different prior distributions for the model parameters when using different learning principles has been overlooked, and possibly lead to questionable conclusions.
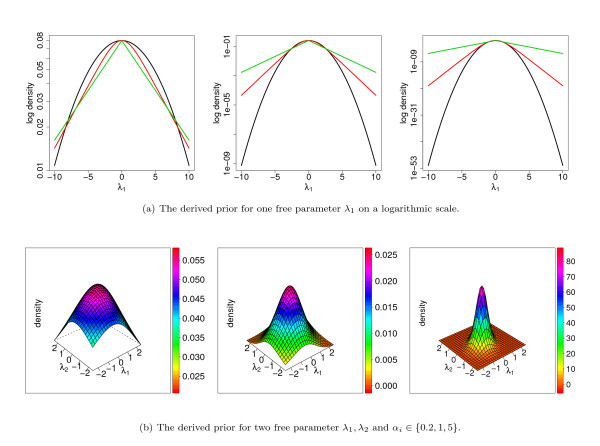
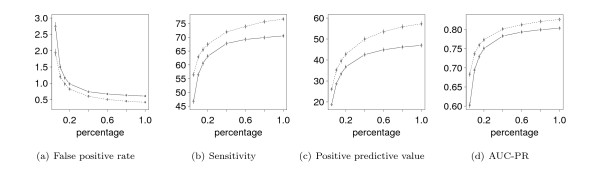
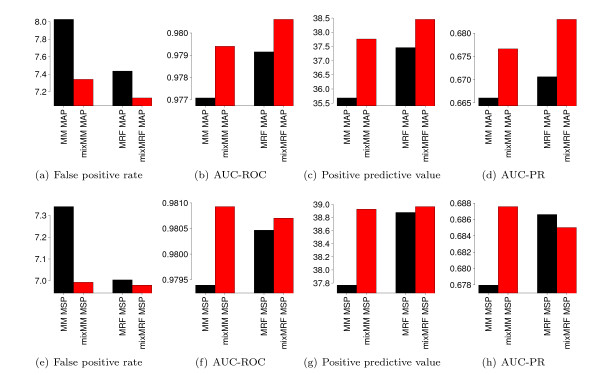
Results: With the goal of allowing direct comparisons of different learning principles for models from the family of Markov random fields based on the same a-priori information, we derive a generalization of the commonly-used product-Dirichlet prior. We find that the derived prior behaves like a Gaussian prior close to the maximum and like a Laplace prior in the far tails. In two case studies, we illustrate the utility of the derived prior for a direct comparison of different learning principles with different models for the recognition of binding sites of the transcription factor Sp1 and human donor splice sites.
Conclusions: We find that comparisons of different learning principles using the same a-priori information can lead to conclusions different from those of previous studies in which the effect resulting from different priors has been neglected. We implement the derived prior is implemented in the open-source library Jstacs to enable an easy application to comparative studies of different learning principles in the field of sequence analysis.
Figures
Similar articles
-
Recognition of cis-regulatory elements with vombat.J Bioinform Comput Biol. 2007 Apr;5(2B):561-77. doi: 10.1142/s0219720007002886. J Bioinform Comput Biol. 2007. PMID: 17636862
-
Transcription binding site prediction using Markov models.J Bioinform Comput Biol. 2006 Apr;4(2):425-41. doi: 10.1142/s0219720006001813. J Bioinform Comput Biol. 2006. PMID: 16819793
-
A transdimensional Bayesian model for pattern recognition in DNA sequences.Biostatistics. 2008 Oct;9(4):668-85. doi: 10.1093/biostatistics/kxm058. Epub 2008 Mar 18. Biostatistics. 2008. PMID: 18349034
-
Hidden Markov model and its applications in motif findings.Methods Mol Biol. 2010;620:405-16. doi: 10.1007/978-1-60761-580-4_13. Methods Mol Biol. 2010. PMID: 20652513 Review.
-
The identification of cis-regulatory elements: A review from a machine learning perspective.Biosystems. 2015 Dec;138:6-17. doi: 10.1016/j.biosystems.2015.10.002. Epub 2015 Oct 21. Biosystems. 2015. PMID: 26499213 Review.
Cited by
-
Varying levels of complexity in transcription factor binding motifs.Nucleic Acids Res. 2015 Oct 15;43(18):e119. doi: 10.1093/nar/gkv577. Epub 2015 Jun 26. Nucleic Acids Res. 2015. PMID: 26116565 Free PMC article.
-
Accurate prediction of cell type-specific transcription factor binding.Genome Biol. 2019 Jan 10;20(1):9. doi: 10.1186/s13059-018-1614-y. Genome Biol. 2019. PMID: 30630522 Free PMC article.
-
A general approach for discriminative de novo motif discovery from high-throughput data.Nucleic Acids Res. 2013 Nov;41(21):e197. doi: 10.1093/nar/gkt831. Epub 2013 Sep 20. Nucleic Acids Res. 2013. PMID: 24057214 Free PMC article.
-
Systems biology data analysis methodology in pharmacogenomics.Pharmacogenomics. 2011 Sep;12(9):1349-60. doi: 10.2217/pgs.11.76. Pharmacogenomics. 2011. PMID: 21919609 Free PMC article. Review.
-
New Algorithm and Software (BNOmics) for Inferring and Visualizing Bayesian Networks from Heterogeneous Big Biological and Genetic Data.J Comput Biol. 2017 Apr;24(4):340-356. doi: 10.1089/cmb.2016.0100. Epub 2016 Sep 28. J Comput Biol. 2017. PMID: 27681505 Free PMC article.
References
-
- Barash Y, Elidan G, Friedman N, Kaplan T. RECOMB '03: Proceedings of the seventh annual international conference on Research in computational molecular biology. New York, NY, USA: ACM Press; 2003. Modelling dependencies in protein-DNA binding sites; pp. 28–37. full_text.
-
- Salzberg SL. A method for identifying splice sites and translational start sites in eukaryotic mRNA. Comput Appl Biosci. 1997;13(4):365–376. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
