

Fast proteomic protocol for biomarker fingerprinting in cancerous cells
- PMID: 20307887
- PMCID: PMC2856699
- DOI: 10.1016/j.chroma.2010.02.065
Fast proteomic protocol for biomarker fingerprinting in cancerous cells
Abstract
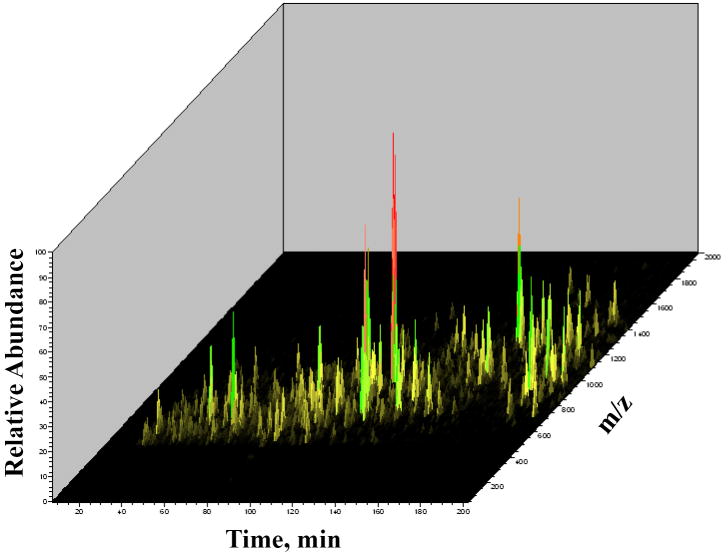
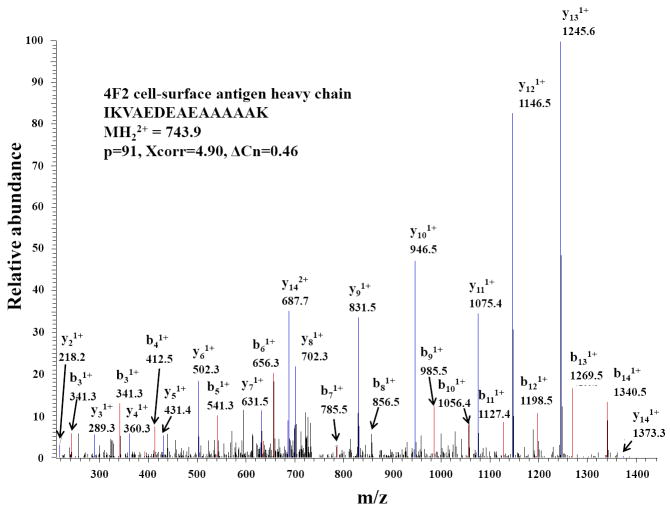
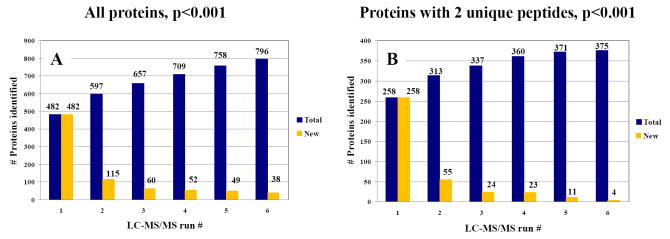
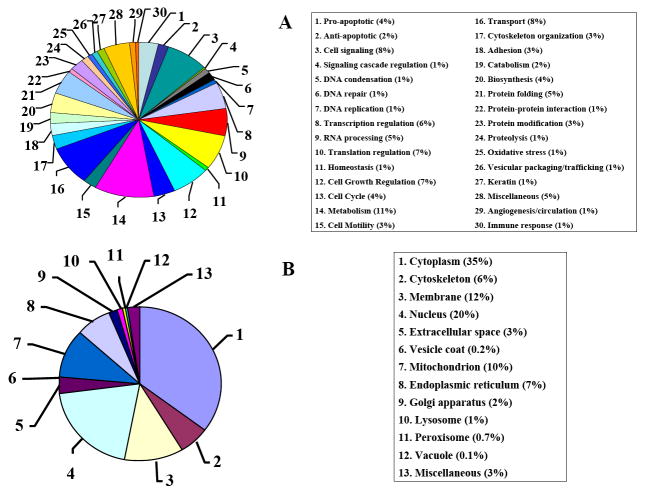
The advance of novel technologies that will enable the detection of large sets of biomarker proteins, to greatly improve the sensitivity and specificity of an assay, represents a major objective in biomedical research. To demonstrate the power of mass spectrometry (MS) detection for large-scale biomarker screening in cancer research, a simple, one-step approach for fast biomarker fingerprinting in complex cellular extracts is described. MCF-7 breast cancer cells were used as a model system. Fast proteomic profiling of whole cellular extracts was achieved on a linear trap quadrupole (LTQ) mass spectrometer by one of the following techniques: (a) data-dependent liquid chromatography (LC)-MS/MS of un-labeled cell extracts, (b) data-dependent LC-MS/MS with pulsed Q dissociation (PQD) detection of iTRAQ labeled samples, and (c) multiple reaction monitoring (MRM)-MS of low abundant proteins that could not be detected with data-dependent MS/MS. The data-dependent LC-MS/MS analysis of MCF-7 cells enabled the identification of 796 proteins (p<0.001) and the simultaneous detection of 156 previously reported putative cancer biomarkers. PQD detection of iTRAQ labeled cells resulted in the detection of 389 proteins and 64 putative biomarkers. MRM-MS analysis enabled the successful monitoring of a panel of low-abundance proteins in one single experiment, highlighting the utility of this technique for targeted analysis in cancer investigations. These results demonstrate that MS-based technologies relying on a one-step separation protocol have the potential to revolutionize biomarker research and screening applications by enabling fast, sensitive and reliable detection of large panels of putative biomarkers. To further stimulate the exploration of proteins that have been previously reported in the literature to be differentially expressed in a variety of cancers, an extensive list of approximately 1100 candidate biomarkers has been compiled and included in the manuscript.
Copyright 2010 Elsevier B.V. All rights reserved.
Figures
Similar articles
-
MRM screening/biomarker discovery with linear ion trap MS: a library of human cancer-specific peptides.BMC Cancer. 2009 Mar 27;9:96. doi: 10.1186/1471-2407-9-96. BMC Cancer. 2009. PMID: 19327145 Free PMC article.
-
Differential protein expression analysis using stable isotope labeling and PQD linear ion trap MS technology.J Am Soc Mass Spectrom. 2009 Jul;20(7):1287-302. doi: 10.1016/j.jasms.2009.02.029. Epub 2009 Mar 4. J Am Soc Mass Spectrom. 2009. PMID: 19345114
-
Proteome profile of the MCF7 cancer cell line: a mass spectrometric evaluation.Rapid Commun Mass Spectrom. 2006;20(20):3039-55. doi: 10.1002/rcm.2677. Rapid Commun Mass Spectrom. 2006. PMID: 16986208
-
Screening of synthetic PDE-5 inhibitors and their analogues as adulterants: analytical techniques and challenges.J Pharm Biomed Anal. 2014 Jan;87:176-90. doi: 10.1016/j.jpba.2013.04.037. Epub 2013 May 6. J Pharm Biomed Anal. 2014. PMID: 23721687 Review.
-
Androgen glucuronides analysis by liquid chromatography tandem-mass spectrometry: could it raise new perspectives in the diagnostic field of hormone-dependent malignancies?J Chromatogr B Analyt Technol Biomed Life Sci. 2013 Dec 1;940:24-34. doi: 10.1016/j.jchromb.2013.09.022. Epub 2013 Sep 27. J Chromatogr B Analyt Technol Biomed Life Sci. 2013. PMID: 24140653 Review.
Cited by
-
Fast Enzymatic Processing of Proteins for MS Detection with a Flow-through Microreactor.J Vis Exp. 2016 Apr 6;(110):e53564. doi: 10.3791/53564. J Vis Exp. 2016. PMID: 27078683 Free PMC article.
-
Microfluidic LC device with orthogonal sample extraction for on-chip MALDI-MS detection.Lab Chip. 2013 Jun 7;13(11):2055-65. doi: 10.1039/c3lc50190f. Lab Chip. 2013. PMID: 23592150 Free PMC article.
-
Quantification of beta-catenin signaling components in colon cancer cell lines, tissue sections, and microdissected tumor cells using reaction monitoring mass spectrometry.J Proteome Res. 2010 Aug 6;9(8):4215-27. doi: 10.1021/pr1005197. J Proteome Res. 2010. PMID: 20590165 Free PMC article.
-
Cell Cycle Model System for Advancing Cancer Biomarker Research.Sci Rep. 2017 Dec 21;7(1):17989. doi: 10.1038/s41598-017-17845-6. Sci Rep. 2017. PMID: 29269772 Free PMC article.
References
-
- Smith L, Lind MJ, Welham KJ, Cawkwell L. Cancer. 2006;107:232. - PubMed
-
- Kumar S, Mohan A, Guleria R. Biomarkers. 2006;11:385. - PubMed
-
- He Q-Y, Chiu J-F. J Cell Biochem. 2003;89:868. - PubMed
-
- Srinivas PR, Verma M, Zhao Y, Srivastava S. Clin Chem. 2002;48:1160. - PubMed
-
- Vlahou A, Fountoulakis M. J Chromatogr B. 2005;814:11. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
