

Long chain acyl-CoA synthetase-3 is a molecular target for peroxisome proliferator-activated receptor delta in HepG2 hepatoma cells
- PMID: 20308079
- PMCID: PMC2878065
- DOI: 10.1074/jbc.M110.112805
Long chain acyl-CoA synthetase-3 is a molecular target for peroxisome proliferator-activated receptor delta in HepG2 hepatoma cells
Abstract
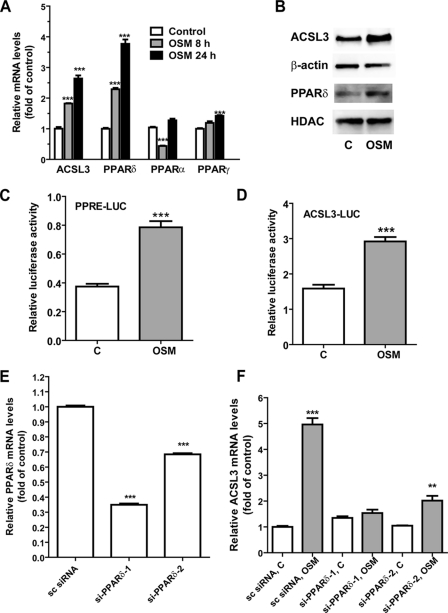
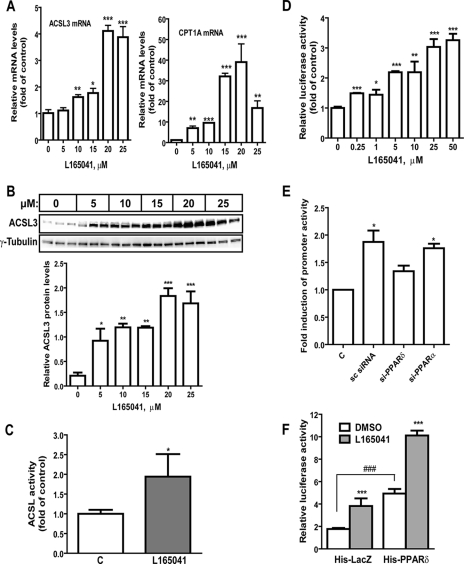
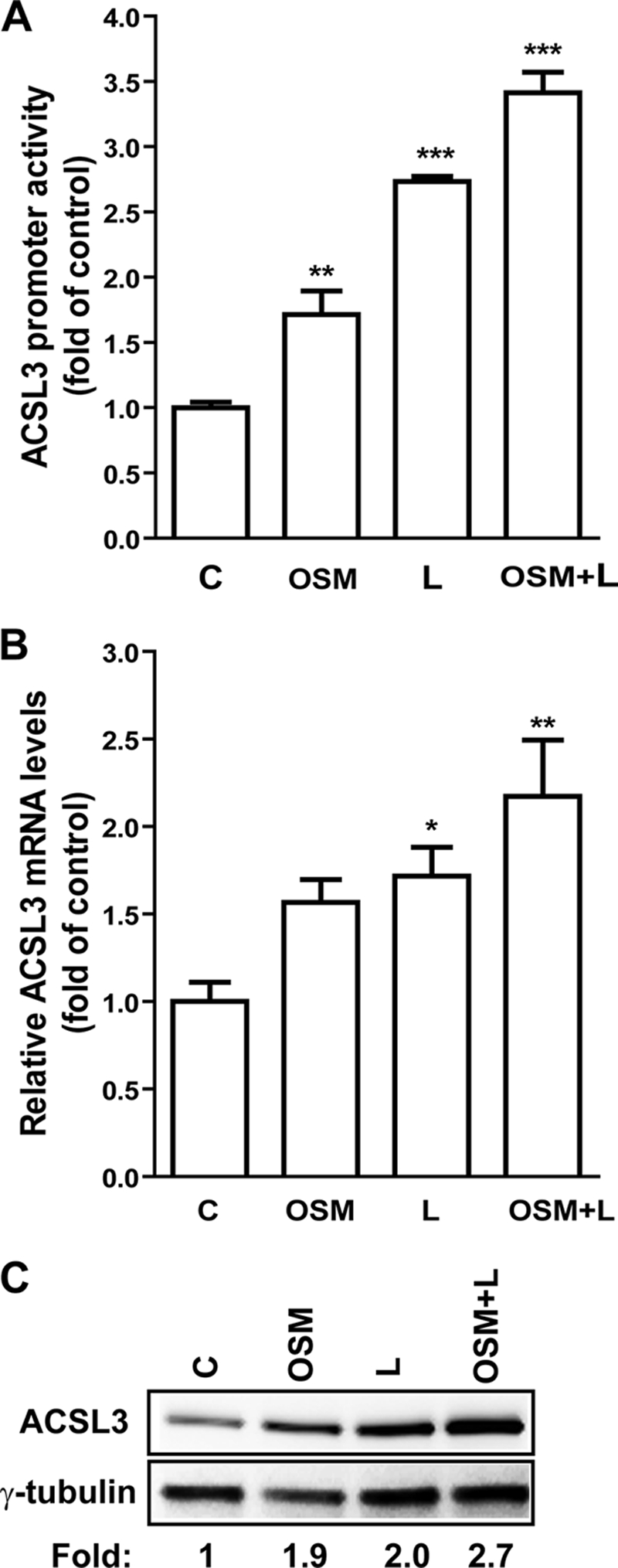
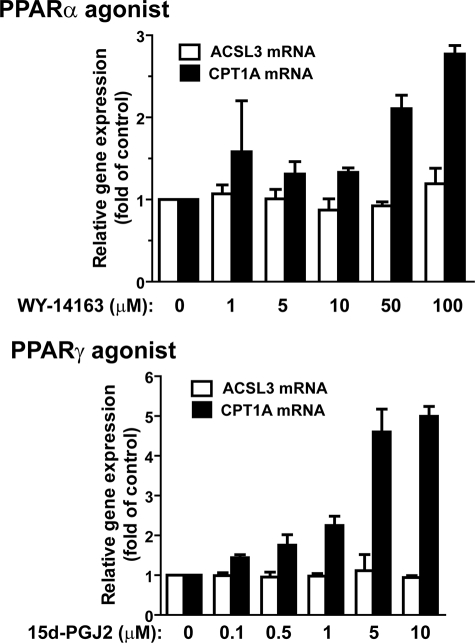
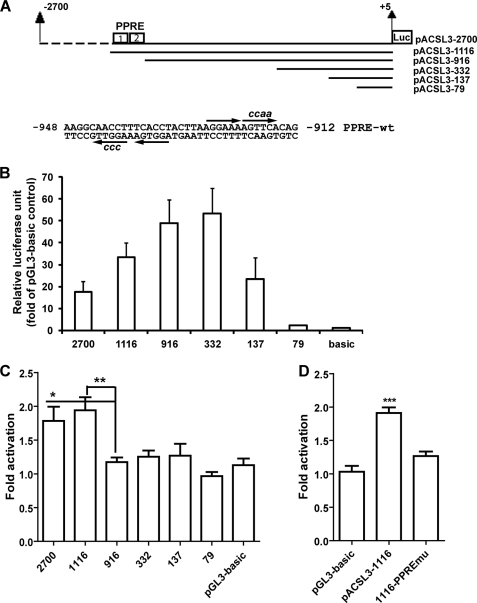
ACSL3 is a member of the long chain acyl-CoA synthetase (ACSL) family that plays key roles in fatty acid metabolism in various tissues in an isozyme-specific manner. Our previous studies showed that ACSL3 was transcriptionally up-regulated by the cytokine oncostatin M (OSM) in HepG2 cells, accompanied by reduced cellular triglyceride content and enhanced beta-oxidation. In this study, we investigated the molecular mechanism underlying the OSM-induced activation of ACSL3 gene transcription in HepG2 cells. We showed that OSM treatment resulted in a coordinated elevation of mRNA levels of ACSL3 and peroxisome proliferator-activated receptor delta (PPARdelta). The effect of OSM on ACSL3 mRNA expression was inhibited by cellular depletion of PPARdelta. By utilizing a PPARdelta agonist, L165041, we demonstrated that activation of PPARdelta led to increases in ACSL3 promoter activity, mRNA level, and protein level in HepG2 cells. Analysis of the ACSL3 promoter sequence identified two imperfect PPAR-responsive elements (PPRE) located in the ACSL3 promoter region -944 to -915, relative to the transcription start site. The up-regulation of ACSL3 promoter activity by PPARdelta was abolished by deletion of this PPRE-containing region or mutation to disrupt the binding sites. Direct interactions of PPARdelta with ACSL3-PPRE sequences were demonstrated by gel mobility shift and chromatin immunoprecipitation assays. Finally, we provided in vivo evidence showing that activation of PPARdelta by L165041 in hamsters increased ACSL3 mRNA and protein levels in the liver. These new findings define ACSL3 as a novel molecular target of PPARdelta in HepG2 cells and provide a regulatory mechanism for ACSL3 transcription in liver tissue.
Figures


References
-
- Mashek D. G., Coleman R. A. (2006) Curr. Opin. Lipidol. 17, 274–278 - PubMed
-
- Suzuki H., Kawarabayasi Y., Kondo J., Abe T., Nishikawa K., Kimura S., Hashimoto T., Yamamoto T. (1990) J. Biol. Chem. 265, 8681–8685 - PubMed
-
- Oikawa E., Iijima H., Suzuki T., Sasano H., Sato H., Kamataki A., Nagura H., Kang M. J., Fujino T., Suzuki H., Yamamoto T. T. (1998) J. Biochem. 124, 679–685 - PubMed
-
- Fujino T., Kang M. J., Suzuki H., Iijima H., Yamamoto T. (1996) J. Biol. Chem. 271, 16748–16752 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
