

Revealing strengths and weaknesses of methods for gene network inference
- PMID: 20308593
- PMCID: PMC2851985
- DOI: 10.1073/pnas.0913357107
Revealing strengths and weaknesses of methods for gene network inference
Abstract
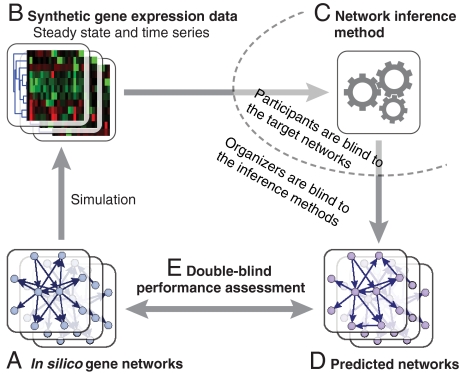
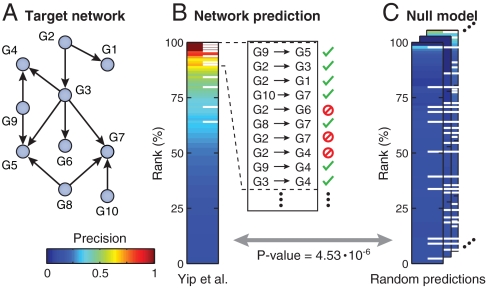
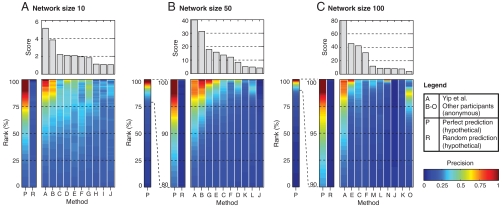
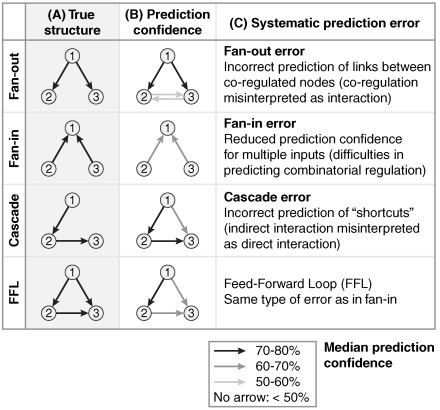
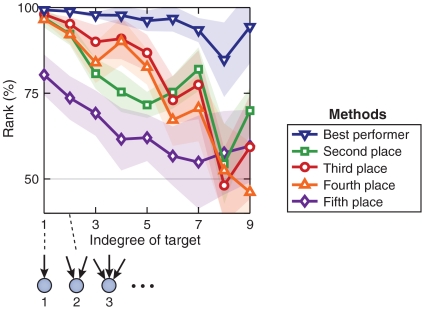
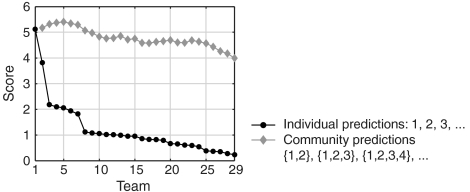
Numerous methods have been developed for inferring gene regulatory networks from expression data, however, both their absolute and comparative performance remain poorly understood. In this paper, we introduce a framework for critical performance assessment of methods for gene network inference. We present an in silico benchmark suite that we provided as a blinded, community-wide challenge within the context of the DREAM (Dialogue on Reverse Engineering Assessment and Methods) project. We assess the performance of 29 gene-network-inference methods, which have been applied independently by participating teams. Performance profiling reveals that current inference methods are affected, to various degrees, by different types of systematic prediction errors. In particular, all but the best-performing method failed to accurately infer multiple regulatory inputs (combinatorial regulation) of genes. The results of this community-wide experiment show that reliable network inference from gene expression data remains an unsolved problem, and they indicate potential ways of network reconstruction improvements.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- De la Fuente A, Brazhnik P, Mendes P. Linking the genes: Inferring quantitative gene networks from microarray data. Trends Genet. 2002;18:395–98. - PubMed
-
- Gardner TS, di Bernardo D, Lorenz D, Collins JJ. Inferring genetic networks and identifying compound mode of action via expression profiling. Science. 2003;301:102–105. - PubMed
-
- Friedman N. Inferring cellular networks using probabilistic graphical models. Science. 2004;303:799–805. - PubMed
-
- Di Bernardo D, et al. Chemogenomic profiling on a genome-wide scale using reverse-engineered gene networks. Nat Biotechnol. 2005;23:377–83. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
