

Review
doi: 10.1038/nrm2873.
Prion-like transmission of protein aggregates in neurodegenerative diseases
Affiliations
- PMID: 20308987
- PMCID: PMC2892479
- DOI: 10.1038/nrm2873
Item in Clipboard
Review
Prion-like transmission of protein aggregates in neurodegenerative diseases
Nat Rev Mol Cell Biol.
2010 Apr.
Abstract
Neurodegenerative diseases are commonly associated with the accumulation of intracellular or extracellular protein aggregates. Recent studies suggest that these aggregates are capable of crossing cellular membranes and can directly contribute to the propagation of neurodegenerative disease pathogenesis. We propose that, once initiated, neuropathological changes might spread in a 'prion-like' manner and that disease progression is associated with the intercellular transfer of pathogenic proteins. The transfer of naked infectious particles between cells could therefore be a target for new disease-modifying therapies.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
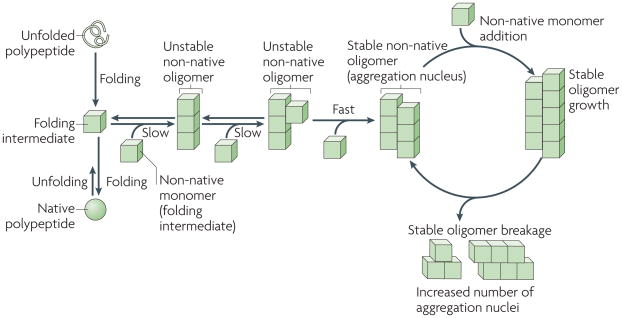
The folding of newly synthesized polypeptide chains into their native conformations and the unfolding of proteins from their native states proceeds through distinct intermediates. some of these intermediates are able to self-associate to form non-native oligomeric species of different sizes and structures, in which a given molecule interacts through two interfaces with two neighbouring molecules (an intermediate in which longitudinal interactions are established is shown). As the polypeptides involved in prion, Parkinson’s, Alzheimer’s and Huntington’s diseases populate a wide variety of folding intermediates, they have a higher propensity to form such oligomeric species. The stability of these oligomers increases on establishment of supplementary intermolecular interactions with non-native polypeptides through additional interfaces, as a given molecule in the oligomer becomes multivalent. The rate-limiting step in non-native polypeptide aggregation is therefore the formation of stable oligomers. Such oligomers behave as nuclei and grow from their ends by recruiting non-native monomers. As the binding of a molecule to the oligomer generates an incorporation site for another subunit, the growth of the stable nuclei is unlimited. Brownian movement and severing and/or disaggregating factors generate increased numbers of ends and increase the likelihood of new subunits being incorporated.
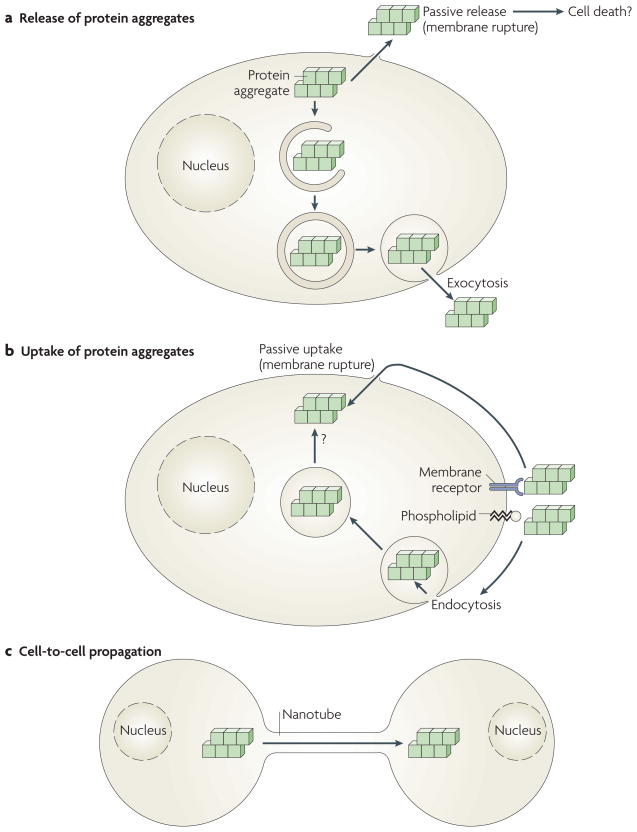
a | Protein aggregates formed in cells can be passively released from cells by membrane rupture or damage, perhaps accompanying cell death. Alternatively, cytoplasmic aggregates can be actively released by exocytosis, possibly following capture by macroautophagy and mis-sorting or incomplete digestion in the endosomal and lysosomal systems. b | Likewise, protein aggregates that bind passively to cell membrane components (phospholipids or protein receptors) can enter the cell either passively by physical rupture of the plasma membrane or actively through endocytosis. Aggregates taken up by endocytosis must cross a biological membrane to reach the cytoplasm, where they can elongate by incorporation of their constituting proteins. c | Protein aggregates, either formed in cells or taken up by cells, can also actively propagate from cell to cell, possibly by cytoskeletal components such as molecular motors and nanotubes (see also BOX 1).
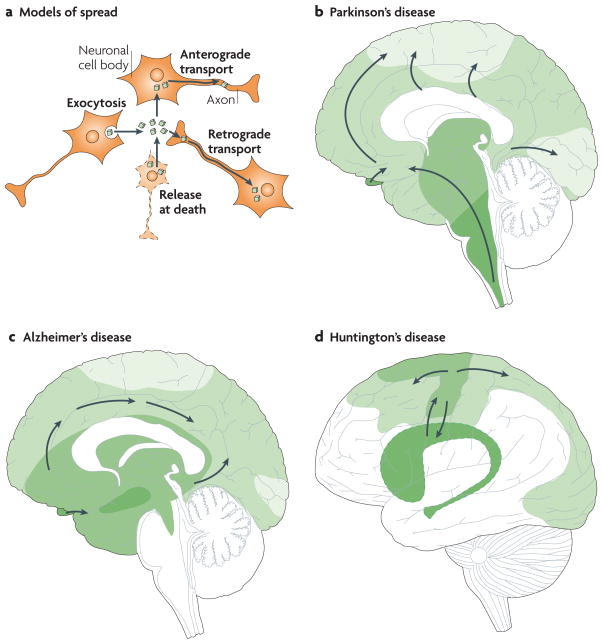
a | Intracellular protein aggregates can be released from neurons by exocytosis or cell death. The aggregates are taken up by, for example, adjacent neuronal cell bodies and are either retained in the cell soma (local spread of pathology) or transported anterogradely by axons. Alternatively, they are taken up by axon terminals and transported retrogradely to the cell soma. The protein aggregates can spread between brain regions by axonal transport. b–d | Three drawings propose principles for how neuropathological changes in Parkinson’s, Alzheimer’s and Huntington’s diseases spread spatiotemporally during disease progression. The earlier the neuropathology develops in a given brain region, the darker the shading in the diagram. As only one view (mid-sagittal for Parkinson’s and Alzheimer’s diseases; lateral for Huntington’s disease) of the brain is depicted for each disorder, not all relevant anatomical structures and details of the spreading patterns (indicated by arrows) are presented. b | in Parkinson’s disease, α-synuclein aggregates (Lewy neurites and Lewy bodies) are suggested to first appear in the dorsal motor nucleus of the vagal nerve in the brainstem and anterior olfactory structures (darkest green), and then to spread stereotypically to finally occupy large parts of the brain,. c | in Alzheimer’s disease, neurofibrillary tangles first appear in the hippocampus (and closely associated structures), the basal nucleus of Meynert and the brainstem– (darkest green). They spread to other brain regions, including the neocortex, in a stereotypical manner, correlating with symptomatic progression. d | in Huntington’s disease, the putamen and caudate nucleus, and related basal ganglia structures deep inside the brain (darkest green), have been suggested to degenerate first–. However, recent imaging studies suggest that primary motor and sensory cortices already undergo atrophy in pre-symptomatic gene carriers. Therefore we propose that cortical involvement precedes basal ganglia pathology.
References
-
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144. - PubMed
-
- Uversky VN. Neuropathology, biochemistry, and biophysics of α-synuclein aggregation. J Neurochem. 2007;103:17–37. - PubMed
-
- Braak H, Ghebremedhin E, Rub U, Bratzke H, Del Tredici K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004;318:121–134. - PubMed
-
- Braak H, et al. Stanley Fahn Lecture 2005: The staging procedure for the inclusion body pathology associated with sporadic Parkinson’s disease reconsidered. Mov Disord. 2006;21:2042–2051. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
