

Novel USH2A compound heterozygous mutations cause RP/USH2 in a Chinese family
- PMID: 20309401
- PMCID: PMC2842093
- DOI: 10.1167/3.9.454
Novel USH2A compound heterozygous mutations cause RP/USH2 in a Chinese family
Abstract
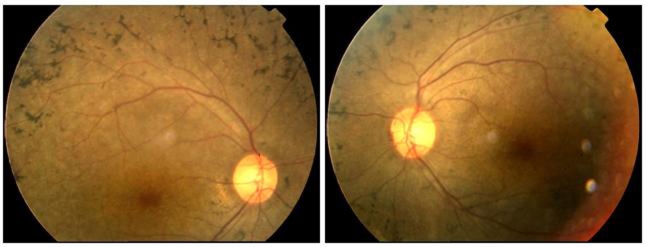
Purpose: To identify the disease-causing gene in a four-generation Chinese family affected with retinitis pigmentosa (RP).
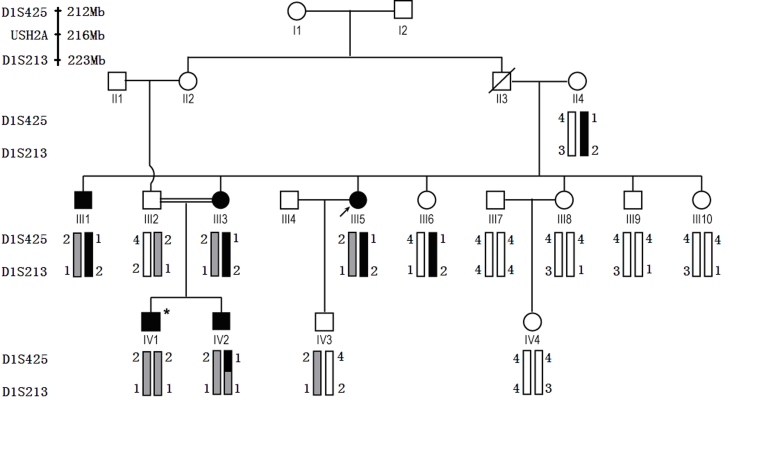
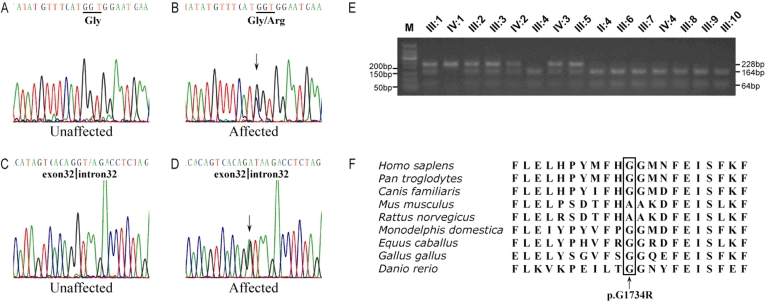
Methods: Linkage analysis was performed with a panel of microsatellite markers flanking the candidate genetic loci of RP. These loci included 38 known RP genes. The complete coding region and exon-intron boundaries of Usher syndrome 2A (USH2A) were sequenced with the proband DNA to screen the disease-causing gene mutation. Restriction fragment length polymorphism (RFLP) analysis and direct DNA sequence analysis were done to demonstrate co-segregation of the USH2A mutations with the family disease. One hundred normal controls were used without the mutations.
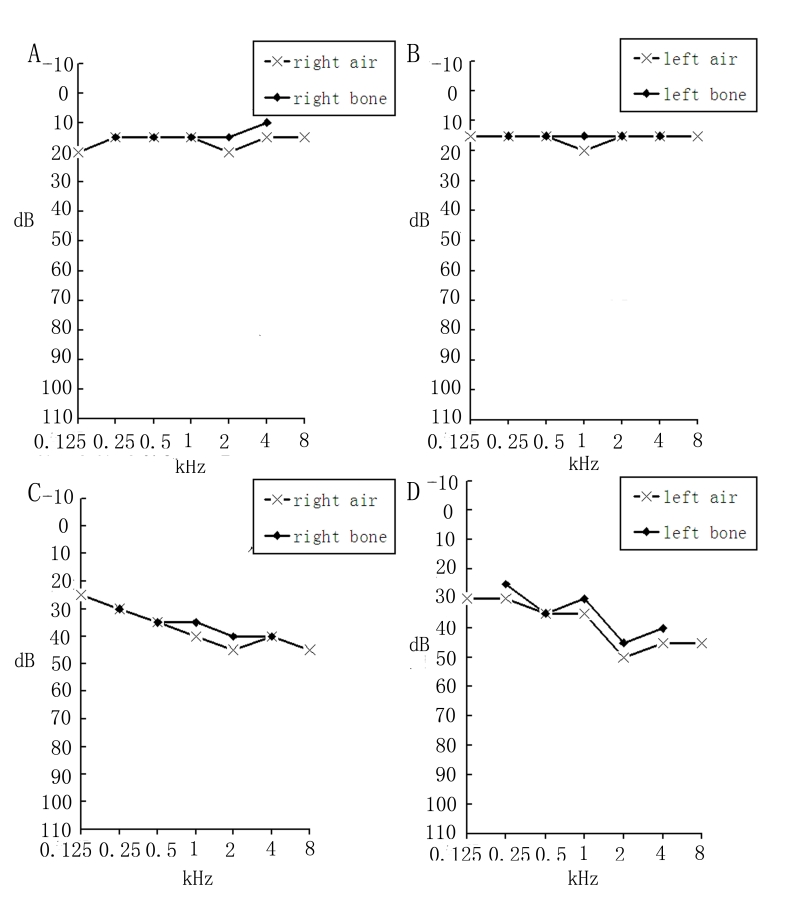
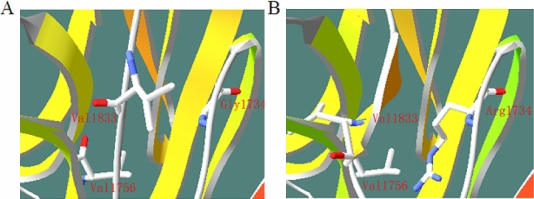
Results: The disease-causing gene in this Chinese family was linked to the USH2A locus on chromosome 1q41. Direct DNA sequence analysis of USH2A identified two novel mutations in the patients: one missense mutation p.G1734R in exon 26 and a splice site mutation, IVS32+1G>A, which was found in the donor site of intron 32 of USH2A. Neither the p.G1734R nor the IVS32+1G>A mutation was found in the unaffected family members or the 100 normal controls. One patient with a homozygous mutation displayed only RP symptoms until now, while three patients with compound heterozygous mutations in the family of study showed both RP and hearing impairment.
Conclusions: This study identified two novel mutations: p.G1734R and IVS32+1G>A of USH2A in a four-generation Chinese RP family. In this study, the heterozygous mutation and the homozygous mutation in USH2A may cause Usher syndrome Type II or RP, respectively. These two mutations expand the mutant spectrum of USH2A.
Figures
Similar articles
-
Seven novel mutations in the long isoform of the USH2A gene in Chinese families with nonsyndromic retinitis pigmentosa and Usher syndrome Type II.Mol Vis. 2011;17:1537-52. Epub 2011 Jun 9. Mol Vis. 2011. PMID: 21686329 Free PMC article.
-
Identification of five novel mutations in the long isoform of the USH2A gene in Chinese families with Usher syndrome type II.Mol Vis. 2008;14:2067-75. Epub 2008 Nov 17. Mol Vis. 2008. PMID: 19023448 Free PMC article.
-
Novel compound heterozygous mutations in MYO7A in a Chinese family with Usher syndrome type 1.Mol Vis. 2013;19:695-701. Epub 2013 Mar 21. Mol Vis. 2013. PMID: 23559863 Free PMC article.
-
Molecular basis of human Usher syndrome: deciphering the meshes of the Usher protein network provides insights into the pathomechanisms of the Usher disease.Exp Eye Res. 2006 Jul;83(1):97-119. doi: 10.1016/j.exer.2005.11.010. Epub 2006 Mar 20. Exp Eye Res. 2006. PMID: 16545802 Review.
-
The p.C759F Variant in USH2A Is a Pathogenic Mutation: Systematic Literature Review and Meta-Analysis of 667 Genotypes.Ophthalmic Res. 2024;67(1):107-114. doi: 10.1159/000535545. Epub 2023 Nov 28. Ophthalmic Res. 2024. PMID: 38016437
Cited by
-
Genotype-phenotype correlations for 17 Chinese families with inherited retinal dystrophies due to homozygous variants.Sci Rep. 2025 Jan 24;15(1):3043. doi: 10.1038/s41598-025-87844-5. Sci Rep. 2025. PMID: 39856360 Free PMC article.
-
Seven novel mutations in the long isoform of the USH2A gene in Chinese families with nonsyndromic retinitis pigmentosa and Usher syndrome Type II.Mol Vis. 2011;17:1537-52. Epub 2011 Jun 9. Mol Vis. 2011. PMID: 21686329 Free PMC article.
-
Exome-wide evidence of compound heterozygous effects across common phenotypes in the UK Biobank.medRxiv [Preprint]. 2023 Jul 3:2023.06.29.23291992. doi: 10.1101/2023.06.29.23291992. medRxiv. 2023. Update in: Cell Genom. 2024 Jul 10;4(7):100602. doi: 10.1016/j.xgen.2024.100602. PMID: 37461573 Free PMC article. Updated. Preprint.
-
The mitotic spindle protein SPAG5/Astrin connects to the Usher protein network postmitotically.Cilia. 2012 Apr 25;1(1):2. doi: 10.1186/2046-2530-1-2. Cilia. 2012. PMID: 23351521 Free PMC article.
-
Exome-wide evidence of compound heterozygous effects across common phenotypes in the UK Biobank.Cell Genom. 2024 Jul 10;4(7):100602. doi: 10.1016/j.xgen.2024.100602. Epub 2024 Jun 28. Cell Genom. 2024. PMID: 38944039 Free PMC article.
References
-
- Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–809. - PubMed
-
- Rosenberg T, Haim M, Hauch AM, Parving A. The prevalence of Usher syndrome and other retinal dystrophy-hearing impairment associations. Clin Genet. 1997;51:314–21. - PubMed
-
- Keats BJ, Corey DP. The usher syndromes. Am J Med Genet. 1999;89:158–66. - PubMed
-
- Smith RJ, Berlin CI, Hejtmancik JF, Keats BJ, Kimberling WJ, Lewis RA, Möller CG, Pelias MZ, Tranebjaerg L. Clinical diagnosis of the Usher syndromes. Usher Syndrome Consortium. Am J Med Genet. 1994;50:32–8. - PubMed
-
- Tsilou ET, Rubin BI, Caruso RC, Reed GF, Pikus A, Hejtmancik JF, Iwata F, Redman JB, Kaiser-Kupfer MI. Usher syndrome clinical types I and II. Acta Ophthalmol Scand. 2002;80:196–201. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases