

TAC3/TACR3 mutations reveal preferential activation of gonadotropin-releasing hormone release by neurokinin B in neonatal life followed by reversal in adulthood
- PMID: 20332248
- PMCID: PMC2902066
- DOI: 10.1210/jc.2009-2320
TAC3/TACR3 mutations reveal preferential activation of gonadotropin-releasing hormone release by neurokinin B in neonatal life followed by reversal in adulthood
Abstract
Context: Mutations in TAC3 and TACR3 (encoding neurokinin B and its receptor) have been identified in Turkish patients with idiopathic hypogonadotropic hypogonadism (IHH), but broader populations have not yet been tested and genotype-phenotype correlations have not been established.
Objective: A broad cohort of normosmic IHH probands was screened for mutations in TAC3/TACR3 to evaluate the prevalence of such mutations and define the genotype/phenotype relationships.
Design and setting: The study consisted of sequencing of TAC3/TACR3, in vitro functional assays, and neuroendocrine phenotyping conducted in tertiary care centers worldwide.
Patients or other participants: 345 probands, 18 family members, and 292 controls were studied.
Intervention: Reproductive phenotypes throughout reproductive life and before and after therapy were examined.
Main outcome measure: Rare sequence variants in TAC3/TACR3 were detected.
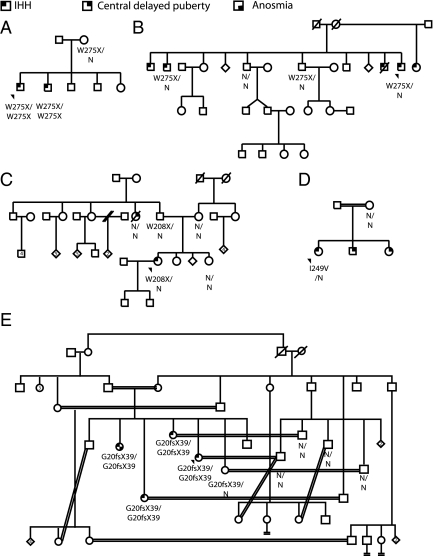
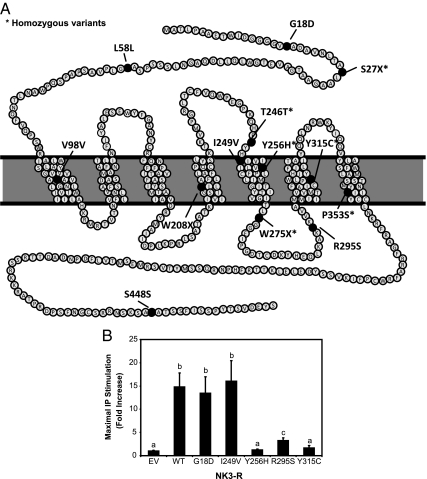
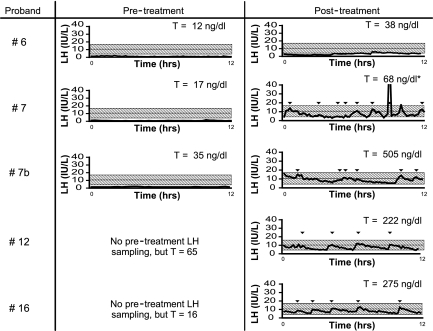
Results: In TACR3, 19 probands harbored 13 distinct coding sequence rare nucleotide variants [three nonsense mutations, six nonsynonymous, four synonymous (one predicted to affect splicing)]. In TAC3, one homozygous single base pair deletion was identified, resulting in complete loss of the neurokinin B decapeptide. Phenotypic information was available on 16 males and seven females with coding sequence variants in TACR3/TAC3. Of the 16 males, 15 had microphallus; none of the females had spontaneous thelarche. Seven of the 16 males and five of the seven females were assessed after discontinuation of therapy; six of the seven males and four of the five females demonstrated evidence for reversibility of their hypogonadotropism.
Conclusions: Mutations in the neurokinin B pathway are relatively common as causes of hypogonadism. Although the neurokinin B pathway appears essential during early sexual development, its importance in sustaining the integrity of the hypothalamic-pituitary-gonadal axis appears attenuated over time.
Figures
Comment in
-
Reversible isolated hypogonadotropic hypogonadism due to mutations in the neurokinin B regulation of gonadotropin-releasing hormone release.J Clin Endocrinol Metab. 2010 Jun;95(6):2625-9. doi: 10.1210/jc.2010-0733. J Clin Endocrinol Metab. 2010. PMID: 20525912 No abstract available.
Similar articles
-
Normosmic congenital hypogonadotropic hypogonadism due to TAC3/TACR3 mutations: characterization of neuroendocrine phenotypes and novel mutations.PLoS One. 2011;6(10):e25614. doi: 10.1371/journal.pone.0025614. Epub 2011 Oct 21. PLoS One. 2011. PMID: 22031817 Free PMC article.
-
TAC3 and TACR3 defects cause hypothalamic congenital hypogonadotropic hypogonadism in humans.J Clin Endocrinol Metab. 2010 May;95(5):2287-95. doi: 10.1210/jc.2009-2600. Epub 2010 Mar 1. J Clin Endocrinol Metab. 2010. PMID: 20194706
-
TAC3 and TACR3 mutations in familial hypogonadotropic hypogonadism reveal a key role for Neurokinin B in the central control of reproduction.Nat Genet. 2009 Mar;41(3):354-358. doi: 10.1038/ng.306. Epub 2008 Dec 11. Nat Genet. 2009. PMID: 19079066 Free PMC article.
-
Neurokinin B signalling in human puberty.J Neuroendocrinol. 2010 Jul;22(7):765-70. doi: 10.1111/j.1365-2826.2010.02013.x. Epub 2010 Apr 29. J Neuroendocrinol. 2010. PMID: 20456599 Review.
-
Neurokinin B and its receptor in hypogonadotropic hypogonadism.Front Horm Res. 2010;39:133-141. doi: 10.1159/000312699. Epub 2010 Apr 8. Front Horm Res. 2010. PMID: 20389091 Review.
Cited by
-
The decline in pulsatile GnRH release, as reflected by circulating LH concentrations, during the infant-juvenile transition in the agonadal male rhesus monkey (Macaca mulatta) is associated with a reduction in kisspeptin content of KNDy neurons of the arcuate nucleus in the hypothalamus.Endocrinology. 2013 May;154(5):1845-53. doi: 10.1210/en.2012-2154. Epub 2013 Mar 22. Endocrinology. 2013. PMID: 23525220 Free PMC article.
-
Reproductive Phenotypes and Genotypes in Men With IHH.J Clin Endocrinol Metab. 2023 Mar 10;108(4):897-908. doi: 10.1210/clinem/dgac615. J Clin Endocrinol Metab. 2023. PMID: 36268624 Free PMC article.
-
Uncovering novel reproductive defects in neurokinin B receptor null mice: closing the gap between mice and men.Endocrinology. 2012 Mar;153(3):1498-508. doi: 10.1210/en.2011-1949. Epub 2012 Jan 17. Endocrinology. 2012. PMID: 22253416 Free PMC article.
-
Mutational analysis of TAC3 and TACR3 genes in patients with idiopathic central pubertal disorders.Arq Bras Endocrinol Metabol. 2012 Dec;56(9):646-52. doi: 10.1590/s0004-27302012000900008. Arq Bras Endocrinol Metabol. 2012. PMID: 23329188 Free PMC article.
-
Do Substance P and Neurokinin A Play Important Roles in the Control of LH Secretion in Ewes?Endocrinology. 2016 Dec;157(12):4829-4841. doi: 10.1210/en.2016-1565. Epub 2016 Oct 5. Endocrinology. 2016. PMID: 27704950 Free PMC article.
References
-
- Franco B, Guioli S, Pragliola A, Incerti B, Bardoni B, Tonlorenzi R, Carrozzo R, Maestrini E, Pieretti M, Taillon-Miller P, Brown CJ, Willard HF, Lawrence C, Persico MG, Camerino G, Ballabio A 1991 A gene deleted in Kallmann's syndrome shares homology with neural cell adhesion and axonal path-finding molecules. Nature 353:529–536 - PubMed
-
- Dodé C, Levilliers J, Dupont JM, De Paepe A, Le Dû N, Soussi-Yanicostas N, Coimbra RS, Delmaghani S, Compain-Nouaille S, Baverel F, Pêcheux C, Le Tessier D, Cruaud C, Delpech M, Speleman F, Vermeulen S, Amalfitano A, Bachelot Y, Bouchard P, Cabrol S, Carel JC, Delemarre-van de Waal H, Goulet-Salmon B, Kottler ML, Richard O, Sanchez-Franco F, Saura R, Young J, Petit C, Hardelin JP 2003 Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet 33:463–465 - PubMed
-
- Falardeau J, Chung WC, Beenken A, Raivio T, Plummer L, Sidis Y, Jacobson-Dickman EE, Eliseenkova AV, Ma J, Dwyer A, Quinton R, Na S, Hall JE, Huot C, Alois N, Pearce SH, Cole LW, Hughes V, Mohammadi M, Tsai P, Pitteloud N 2008 Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice. J Clin Invest 118:2822–2831 - PMC - PubMed
-
- Miura K, Acierno Jr JS, Seminara SB 2004 Characterization of the human nasal embryonic LHRH factor gene, NELF, and a mutation screening among 65 patients with idiopathic hypogonadotropic hypogonadism (IHH). J Hum Genet 49:265–268 - PubMed
-
- Matsumoto S, Yamazaki C, Masumoto KH, Nagano M, Naito M, Soga T, Hiyama H, Matsumoto M, Takasaki J, Kamohara M, Matsuo A, Ishii H, Kobori M, Katoh M, Matsushime H, Furuichi K, Shigeyoshi Y 2006 Abnormal development of the olfactory bulb and reproductive system in mice lacking prokineticin receptor PKR2. Proc Natl Acad Sci USA 103:4140–4145 - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
