

How I treat common variable immune deficiency
- PMID: 20332369
- PMCID: PMC2904582
- DOI: 10.1182/blood-2010-01-254417
How I treat common variable immune deficiency
Abstract
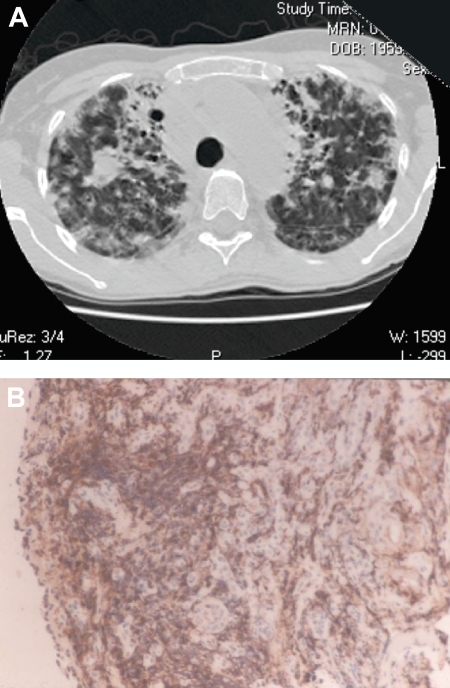
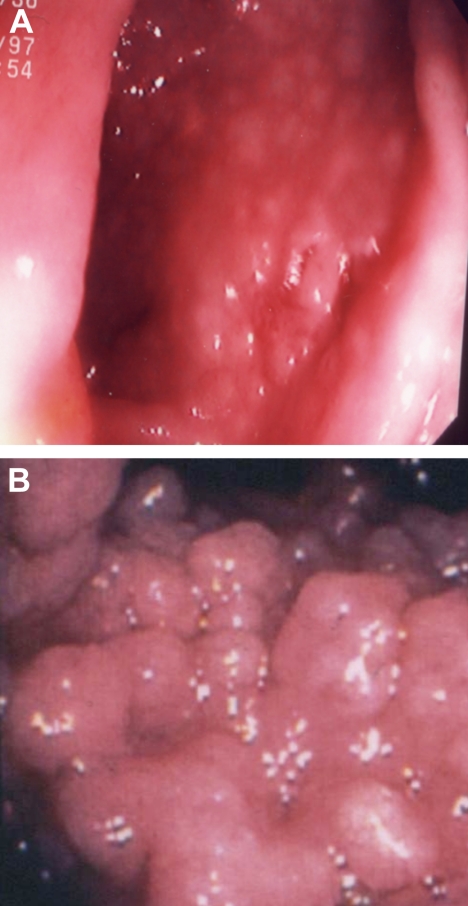
Common variable immunodeficiency is a rare immune deficiency, characterized by low levels of serum immunoglobulin G, A, and/or M with loss of antibody production. The diagnosis is most commonly made in adults between the ages of 20 and 40 years, but both children and older adults can be found to have this immune defect. The range of clinical manifestations is broad, including acute and chronic infections, inflammatory and autoimmune disease, and an increased incidence of cancer and lymphoma. For all these reasons, the disease phenotype is both heterogeneous and complex. Contributing to the complexity is that patient cohorts are generally small, criteria used for diagnosis vary, and the doses of replacement immune globulin differ. In addition, routines for monitoring patients over the years and protocols for the use of other biologic agents for complications have not been clarified or standardized. In the past few years, data from large patient registries have revealed that both selected laboratory markers and clinical phenotyping may aid in dissecting groups of subjects into biologically relevant categories. This review presents my approach to the diagnosis and treatment of patients with common variable immunodeficiency, with suggestions for the use of laboratory biomarkers and means of monitoring patients.
Figures
References
-
- Chapel H, Lucas M, Lee M, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. 2008;112(2):277–286. - PubMed
-
- Janeway CA, Apt L, Gitlin D. Agammaglobulinemia. Trans Assoc Am Phys. 1953;66:200–202. - PubMed
-
- Hermaszewski RA, Webster AD. Primary hypogammaglobulinaemia: a survey of clinical manifestations and complications. Q J Med. 1993;86(1):31–42. - PubMed
-
- Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol. 1999;92(1):34–48. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
