

ER calcium and Alzheimer's disease: in a state of flux
- PMID: 20332425
- PMCID: PMC3091478
- DOI: 10.1126/scisignal.3114pe10
ER calcium and Alzheimer's disease: in a state of flux
Abstract
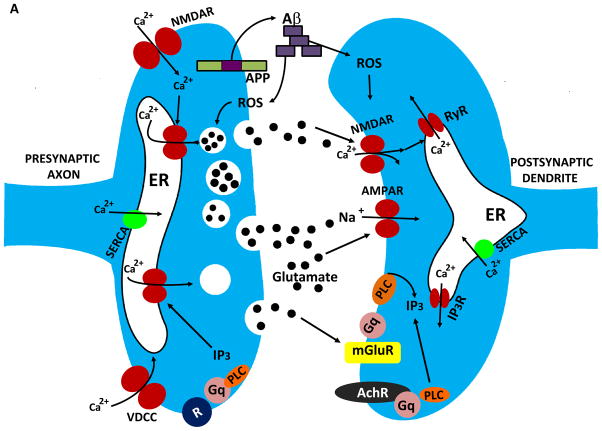
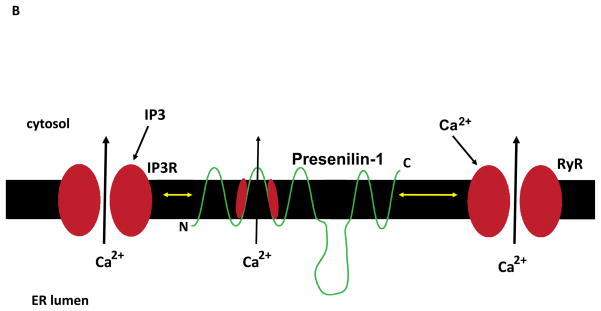
The calcium ion (Ca(2+)) plays fundamental roles in orchestrating dynamic changes in the function and structure of nerve cell circuits in the brain. The endoplasmic reticulum (ER), an organelle that actively removes Ca(2+) from the cytoplasm, can release stored Ca(2+) through ER membrane receptor channels responsive either to the lipid messenger inositol trisphosphate (IP(3)) or to cytosolic Ca(2+). Emerging findings suggest that perturbed ER Ca(2+) homeostasis contributes to the dysfunction and degeneration of neurons that occurs in Alzheimer's disease (AD). Presenilin-1 (PS1) is an integral membrane protein in the ER; mutations in PS1 that cause early-onset inherited AD increase the pool of ER Ca(2+) available for release and also enhance Ca(2+) release through ER IP(3)- and ryanodine-sensitive channels. By enhancing Ca(2+) flux across the ER membrane, PS1 mutations may exaggerate Ca(2+) signaling in synaptic terminals and thereby render them vulnerable to dysfunction and degeneration in the settings of aging and amyloid accumulation in AD.
Figures
References
-
- Alzheimer’s Association. Alzheimer’s disease facts and figures. Alzheimers Dement. 2009;5:234–270. - PubMed
-
- Arumugam TV, Gleichmann M, Tang SC, Mattson MP. Hormesis/preconditioning mechanisms, the nervous system and aging. Ageing Res Rev. 2006;5:165–178. - PubMed
-
- Cohen AD, Price J, Weissfeld L, James J, Rosario B, Bi W, Nebes R, Saxton J, Snitz BE, Aizenstein HA, Wolk DA, Dekosky ST, Mathis CA, Klunk WE. Basal cerebral metabolism may modulate the cognitive effects of Abeta in mild cognitive impairment: an example of brain reserve. J Neurosci. 2009;29:14770–14778. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
