

Cleft palate is caused by CNS dysfunction in Gad1 and Viaat knockout mice
- PMID: 20333300
- PMCID: PMC2841638
- DOI: 10.1371/journal.pone.0009758
Cleft palate is caused by CNS dysfunction in Gad1 and Viaat knockout mice
Abstract
Background: Previous studies have shown that disruption of GABA signaling in mice via mutations in the Gad1, Gabrb3 or Viaat genes leads to the development of non-neural developmental defects such as cleft palate. Studies of the Gabrb3 and Gad1 mutant mice have suggested that GABA function could be required either in the central nervous system or in the palate itself for normal palatogenesis.
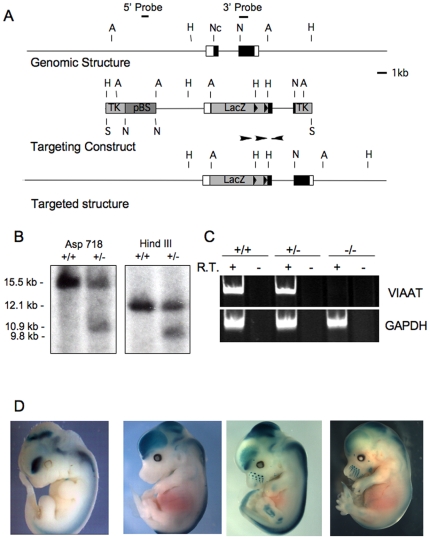
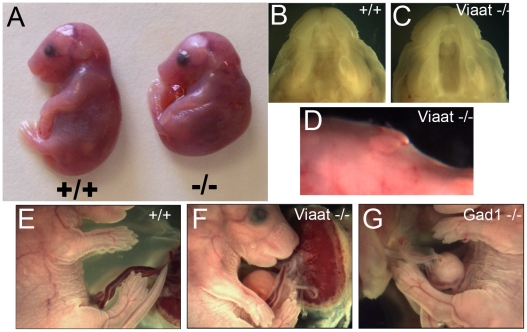
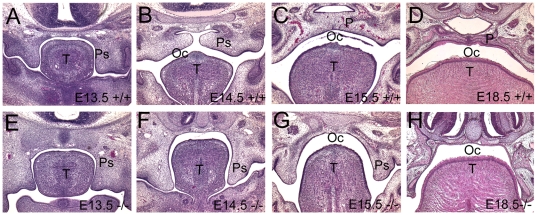
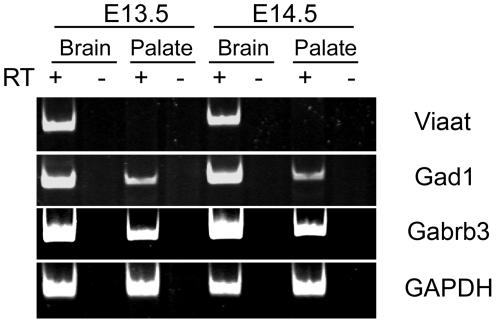
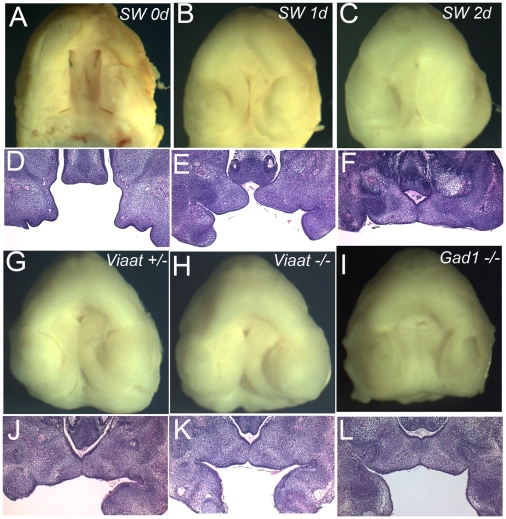
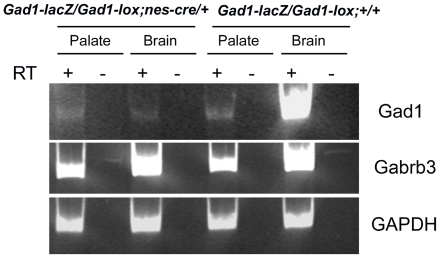
Methodology/principal findings: To further examine the role of GABA signaling in palatogenesis we used three independent experimental approaches to test whether Gad1 or Viaat function is required in the fetal CNS for normal palate development. We used oral explant cultures to demonstrate that the Gad1 and Viaat mutant palates were able to undergo palatogenesis in culture, suggesting that there is no defect in the palate tissue itself in these mice. In a second series of experiments we found that the GABA(A) receptor agonist muscimol could rescue the cleft palate phenotype in Gad1 and Viaat mutant embryos. This suggested that normal multimeric GABA(A) receptors in the CNS were necessary for normal palatogenesis. In addition, we showed that CNS-specific inactivation of Gad1 was sufficient to disrupt palate development.
Conclusions/significance: Our results are consistent with a role for Gad1 and Viaat in the central nervous system for normal development of the palate. We suggest that the alterations in GABA signaling lead to non-neural defects such as cleft palate as a secondary effect due to alterations in or elimination of fetal movements.
Conflict of interest statement
Figures
References
-
- Culiat CT, Stubbs LJ, Woychik RP, Russell LB, Johnson DK, et al. Deficiency of the beta 3 subunit of the type A gamma-aminobutyric acid receptor causes cleft palate in mice. Nat Genet. 1995;11:344–346. - PubMed
-
- Wojcik SM, Katsurabayashi S, Guillemin I, Friauf E, Rosenmund C, et al. A shared vesicular carrier allows synaptic corelease of GABA and glycine. Neuron. 2006;50:575–587. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
