

Reciprocal regulation of metabolic and signaling pathways
- PMID: 20334672
- PMCID: PMC2861677
- DOI: 10.1186/1471-2164-11-197
Reciprocal regulation of metabolic and signaling pathways
Abstract
Background: By studying genome-wide expression patterns in healthy and diseased tissues across a wide range of pathophysiological conditions, DNA microarrays have revealed unique insights into complex diseases. However, the high-dimensionality of microarray data makes interpretation of heterogeneous gene expression studies inherently difficult.
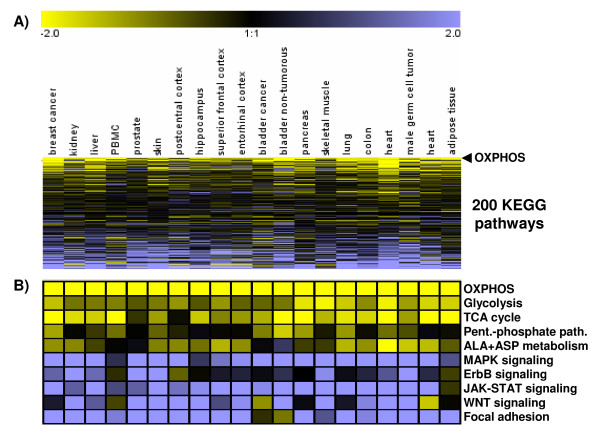
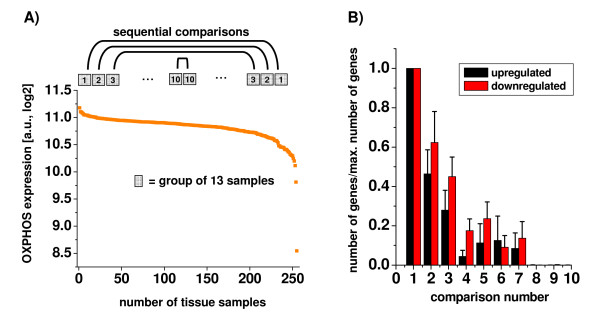
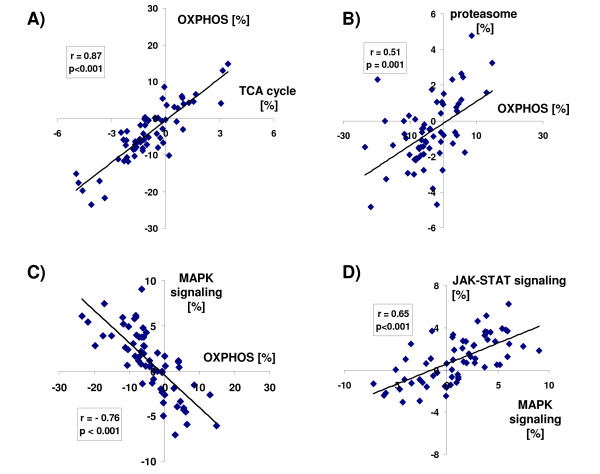
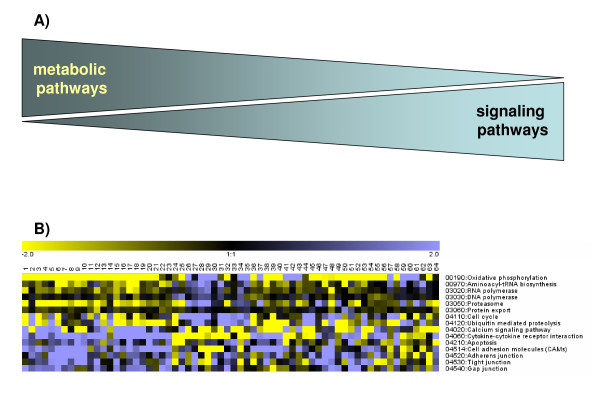
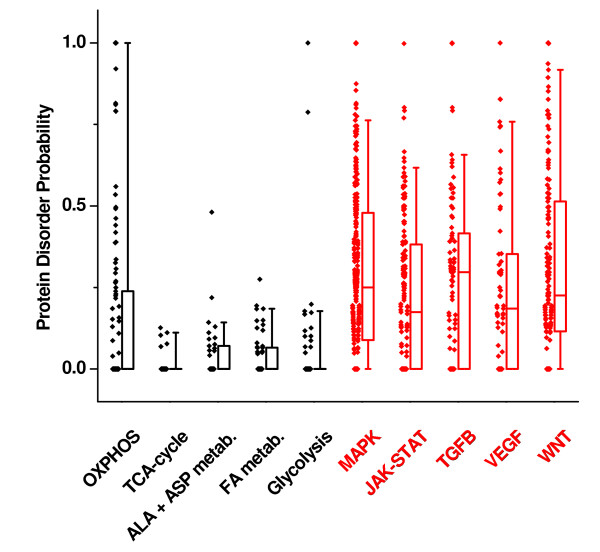
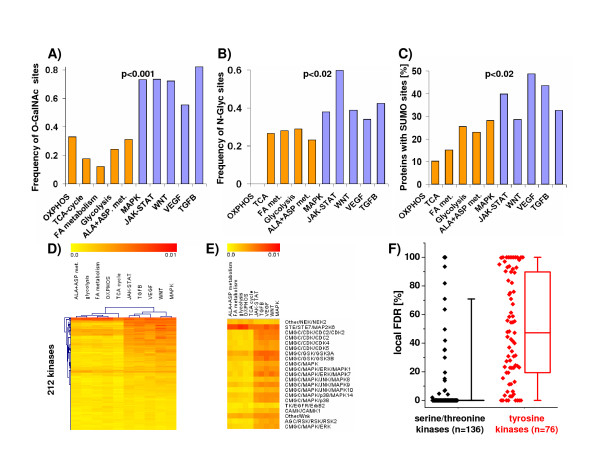
Results: Using a large-scale analysis of more than 40 microarray studies encompassing ~2400 mammalian tissue samples, we identified a common theme across heterogeneous microarray studies evident by a robust genome-wide inverse regulation of metabolic and cell signaling pathways: We found that upregulation of cell signaling pathways was invariably accompanied by downregulation of cell metabolic transcriptional activity (and vice versa). Several findings suggest that this characteristic gene expression pattern represents a new principle of mammalian transcriptional regulation. First, this coordinated transcriptional pattern occurred in a wide variety of physiological and pathophysiological conditions and was identified across all 20 human and animal tissue types examined. Second, the differences in metabolic gene expression predicted the magnitude of differences for signaling and all other pathways, i.e. tissue samples with similar expression levels of metabolic transcripts did not show any differences in gene expression for all other pathways. Third, this transcriptional pattern predicted a profound effect on the proteome, evident by differences in structure, stability and post-translational modifications of proteins belonging to signaling and metabolic pathways, respectively.
Conclusions: Our data suggest that in a wide range of physiological and pathophysiological conditions, gene expression changes exhibit a recurring pattern along a transcriptional axis, characterized by an inverse regulation of major metabolic and cell signaling pathways. Given its widespread occurrence and its predicted effects on protein structure, protein stability and post-translational modifications, we propose a new principle for transcriptional regulation in mammalian biology.
Figures
References
-
- Barth AS, Aiba T, Halperin V, DiSilvestre D, Chakir K, Colantuoni C, Tunin RS, Dimaano VL, Yu W, Abraham TP. et al. Cardiac resynchronization therapy corrects dyssynchrony-induced regional gene expression changes on a genomic level. Circ Cardiovasc Genet. 2009;2(4):371–378. doi: 10.1161/CIRCGENETICS.108.832345. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
