

Quantitative analysis of cryo-EM density map segmentation by watershed and scale-space filtering, and fitting of structures by alignment to regions
- PMID: 20338243
- PMCID: PMC2874196
- DOI: 10.1016/j.jsb.2010.03.007
Quantitative analysis of cryo-EM density map segmentation by watershed and scale-space filtering, and fitting of structures by alignment to regions
Abstract
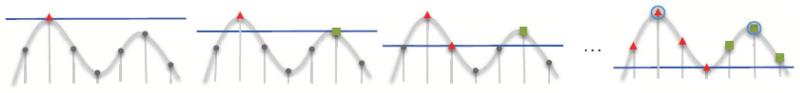
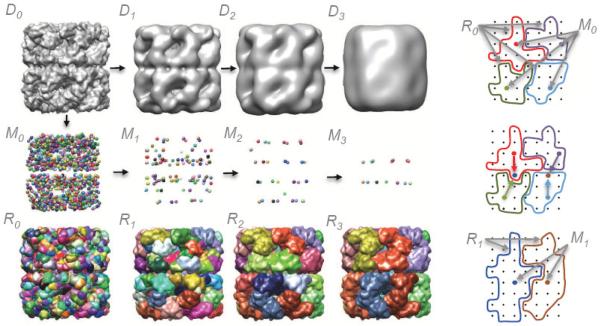
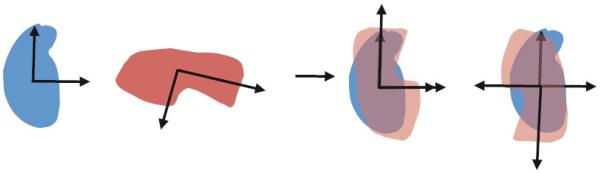
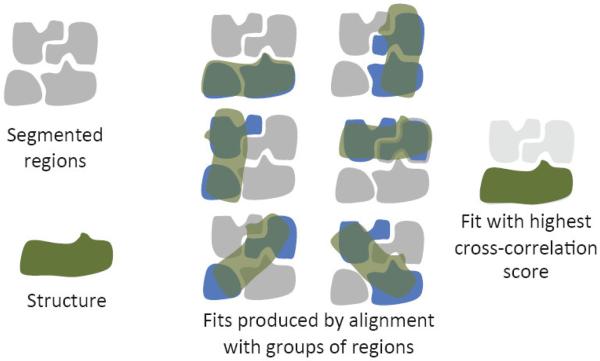
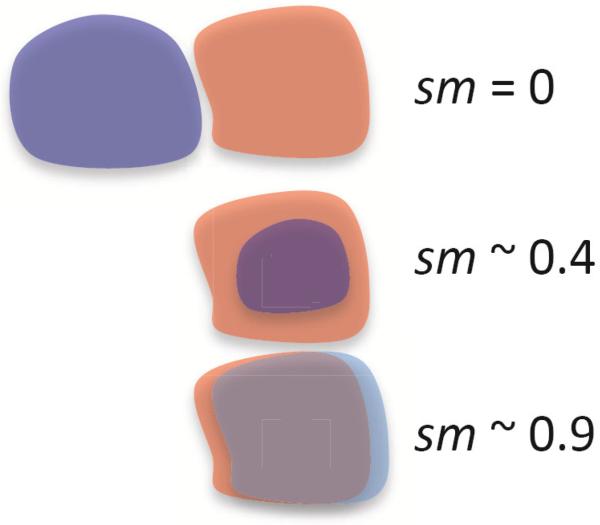
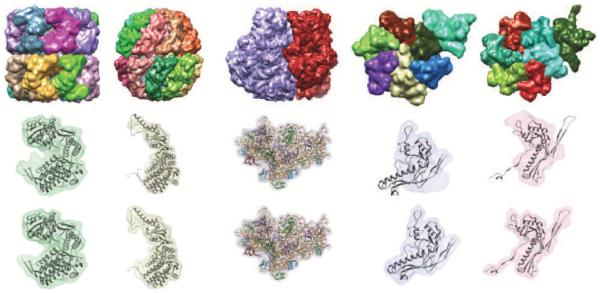
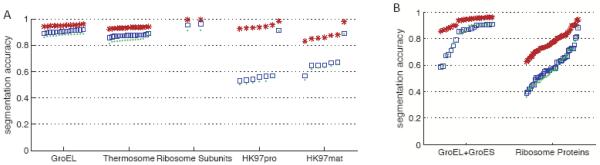
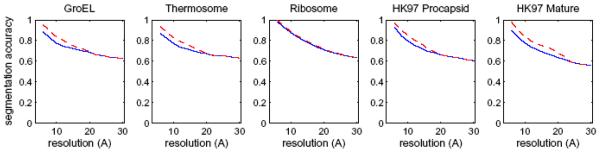
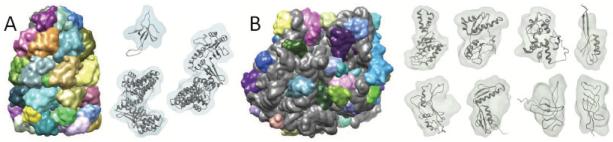
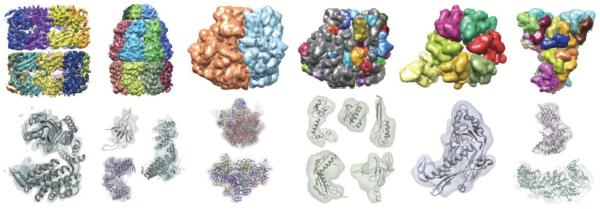
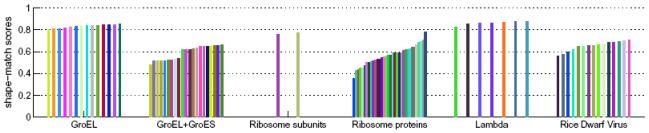
Cryo-electron microscopy produces 3D density maps of molecular machines, which consist of various molecular components such as proteins and RNA. Segmentation of individual components in such maps is a challenging task, and is mostly accomplished interactively. We present an approach based on the immersive watershed method and grouping of the resulting regions using progressively smoothed maps. The method requires only three parameters: the segmentation threshold, a smoothing step size, and the number of smoothing steps. We first apply the method to maps generated from molecular structures and use a quantitative metric to measure the segmentation accuracy. The method does not attain perfect accuracy, however it produces single or small groups of regions that roughly match individual proteins or subunits. We also present two methods for fitting of structures into density maps, based on aligning the structures with single regions or small groups of regions. The first method aligns centers and principal axes, whereas the second aligns centers and then rotates the structure to find the best fit. We describe both interactive and automated ways of using these two methods. Finally, we show segmentation and fitting results for several experimentally-obtained density maps.
Figures
References
-
- Ludtke SJ, Baker ML, Chen D, Song J, Chuang DT, Chiu W. De novo backbone trace of GroEL from single particle electron cryomicroscopy. Structure. 2008;16:441–448. - PubMed
-
- Valle M, Zavialov A, Li W, Stagg SM, Sengupta J, Nielsen RC, et al. Incorporation of aminoacyl-tRNA into the ribosome as seen by cryo-electron microscopy. Nat Struct Mol Biol. 2003;10:899–906. - PubMed
-
- Zhou ZH, Baker ML, Jiang W, Dougherty M, Jakana J, Dong G, et al. Electron cryomicroscopy and bioinformatics suggest protein fold models for rice dwarf virus. Nat Struct Mol Biol. 2001;8:868–873. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
