

CDK-Taverna: an open workflow environment for cheminformatics
- PMID: 20346188
- PMCID: PMC2862046
- DOI: 10.1186/1471-2105-11-159
CDK-Taverna: an open workflow environment for cheminformatics
Abstract
Background: Small molecules are of increasing interest for bioinformatics in areas such as metabolomics and drug discovery. The recent release of large open access chemistry databases generates a demand for flexible tools to process them and discover new knowledge. To freely support open science based on these data resources, it is desirable for the processing tools to be open source and available for everyone.
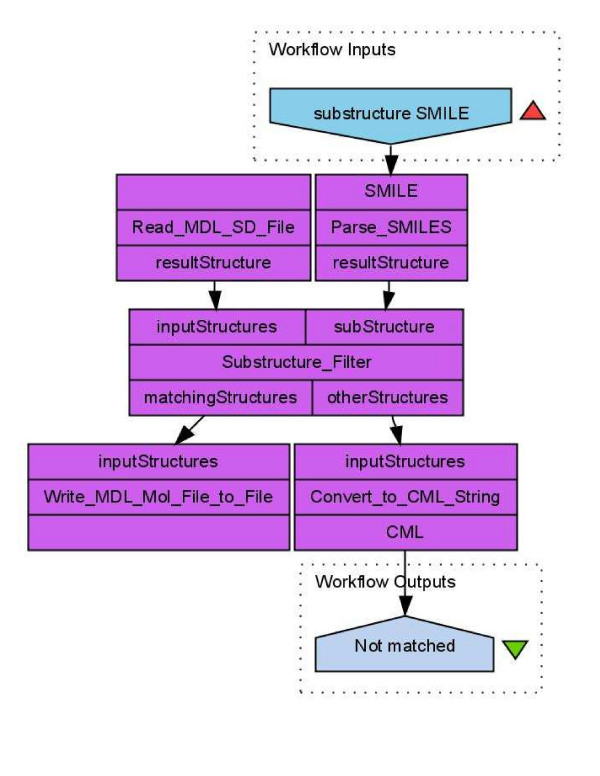
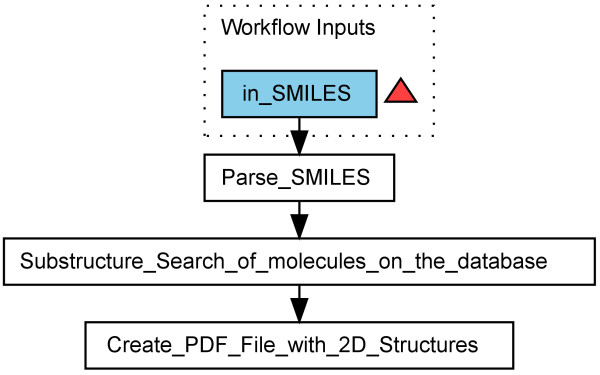
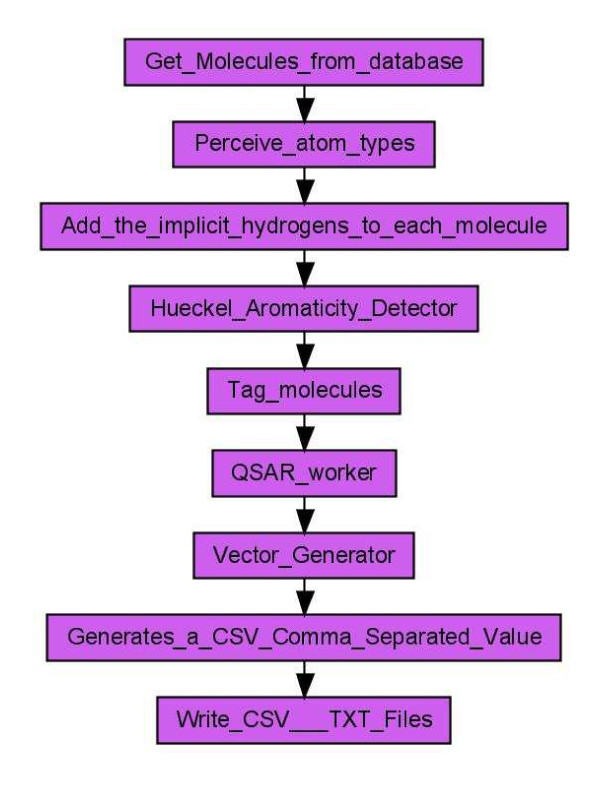
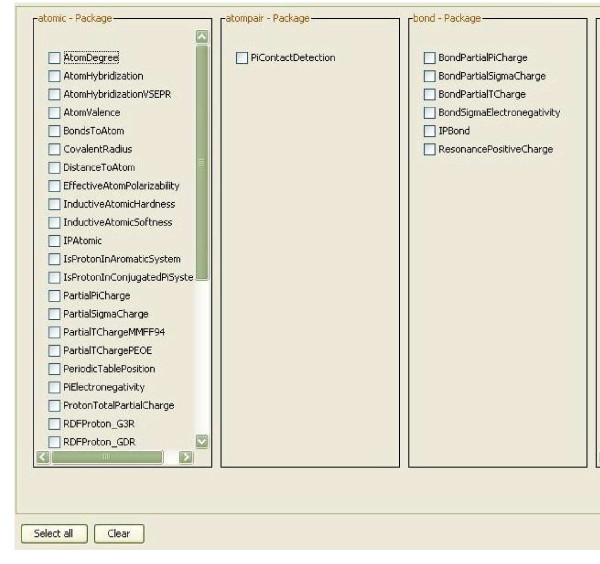
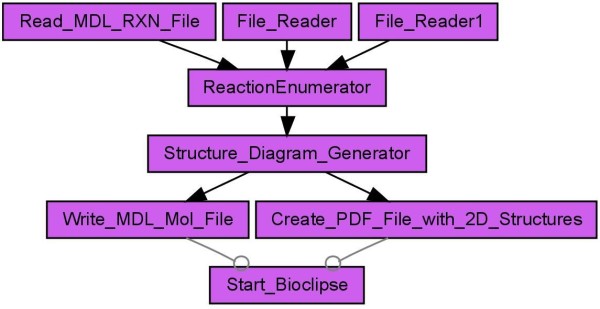
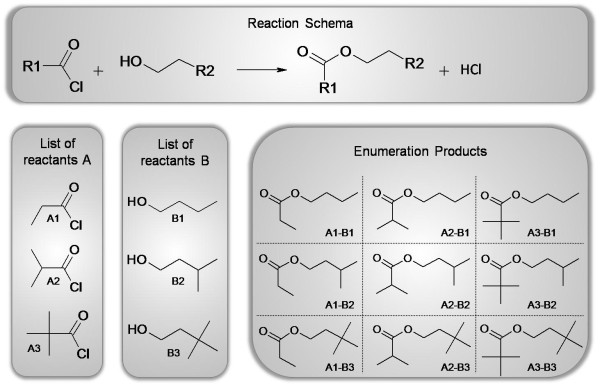
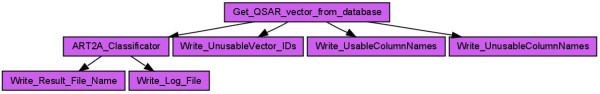
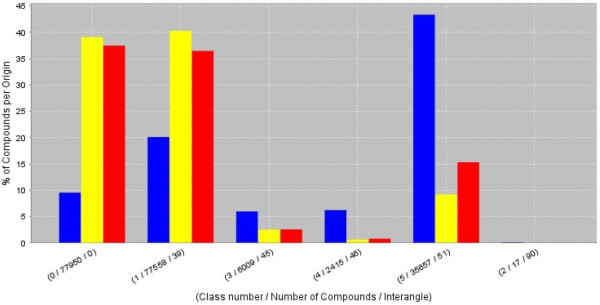
Results: Here we describe a novel combination of the workflow engine Taverna and the cheminformatics library Chemistry Development Kit (CDK) resulting in a open source workflow solution for cheminformatics. We have implemented more than 160 different workers to handle specific cheminformatics tasks. We describe the applications of CDK-Taverna in various usage scenarios.
Conclusions: The combination of the workflow engine Taverna and the Chemistry Development Kit provides the first open source cheminformatics workflow solution for the biosciences. With the Taverna-community working towards a more powerful workflow engine and a more user-friendly user interface, CDK-Taverna has the potential to become a free alternative to existing proprietary workflow tools.
Figures
Similar articles
-
New developments on the cheminformatics open workflow environment CDK-Taverna.J Cheminform. 2011 Dec 13;3:54. doi: 10.1186/1758-2946-3-54. J Cheminform. 2011. PMID: 22166170 Free PMC article.
-
BIFI: a Taverna plugin for a simplified and user-friendly workflow platform.BMC Res Notes. 2014 Oct 20;7:740. doi: 10.1186/1756-0500-7-740. BMC Res Notes. 2014. PMID: 25332013 Free PMC article.
-
The Taverna Interaction Service: enabling manual interaction in workflows.Bioinformatics. 2008 Apr 15;24(8):1118-20. doi: 10.1093/bioinformatics/btn082. Epub 2008 Mar 12. Bioinformatics. 2008. PMID: 18337261
-
Enalos Suite: New Cheminformatics Platform for Drug Discovery and Computational Toxicology.Methods Mol Biol. 2018;1800:287-311. doi: 10.1007/978-1-4939-7899-1_14. Methods Mol Biol. 2018. PMID: 29934899 Review.
-
Workflow based framework for life science informatics.Comput Biol Chem. 2007 Oct;31(5-6):305-19. doi: 10.1016/j.compbiolchem.2007.08.009. Epub 2007 Aug 19. Comput Biol Chem. 2007. PMID: 17931570 Review.
Cited by
-
Towards reproducible computational drug discovery.J Cheminform. 2020 Jan 28;12(1):9. doi: 10.1186/s13321-020-0408-x. J Cheminform. 2020. PMID: 33430992 Free PMC article. Review.
-
Scaffold Hunter: a comprehensive visual analytics framework for drug discovery.J Cheminform. 2017 May 11;9(1):28. doi: 10.1186/s13321-017-0213-3. J Cheminform. 2017. PMID: 29086162 Free PMC article.
-
Natural product-likeness score revisited: an open-source, open-data implementation.BMC Bioinformatics. 2012 May 20;13:106. doi: 10.1186/1471-2105-13-106. BMC Bioinformatics. 2012. PMID: 22607271 Free PMC article.
-
Chembench: a cheminformatics workbench.Bioinformatics. 2010 Dec 1;26(23):3000-1. doi: 10.1093/bioinformatics/btq556. Epub 2010 Sep 30. Bioinformatics. 2010. PMID: 20889496 Free PMC article.
-
Increasing the Value of Data Within a Large Pharmaceutical Company Through In Silico Models.Methods Mol Biol. 2022;2425:637-674. doi: 10.1007/978-1-0716-1960-5_24. Methods Mol Biol. 2022. PMID: 35188649
References
-
- The PubChem Project. http://pubchem.ncbi.nlm.nih.gov/
-
- The ChEMBL Group. http://www.ebi.ac.uk/chembl
-
- Williams AJ. Public chemical compound databases. Current opinion in drug discovery & development. 2008;11(3):393–404. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
